Researchers who packed the lecture halls for “Common Mechanisms of Neurodegeneration” and “Microglia in the Brain,” joint Keystone Symposia held June 12-16 in Keystone, Colorado, saw old dogmas fall and new ideas and methodologies emerge. Debates ran the gamut from the biophysical nature of toxic proteins to the characterization of microglia in health and disease.
Keystone Meeting on Microglia/Neurodegeneration: Here’s the Buzz
Sometimes arriving even 15 minutes early to a lecture hall can turn out to be, well, too late. Interest was so keen in “Common Mechanisms of Neurodegeneration” and “Microglia in the Brain,” joint Keystone Symposia held June 12-16 in Keystone, Colorado, that attendees were claiming seats well before sessions started with their bags, books, computers, and other personal items. “Tardy” folks scrambled for whatever spots were left at the back of packed hallways, or sat on the floor to be close to the screen. Yet no one complained. A spirit of cooperation and collaboration was palpable at this meeting, as researchers seemed united in a common purpose to figure out why neurons degenerate and what role microglia play in that process and indeed the healthy brain. Young and old researchers alike debated well into the night around packed posters, where patience paid off for those trying to squeeze close enough to see the data up-close, and co-authors tag-teamed to present their findings for the umpteenth time.
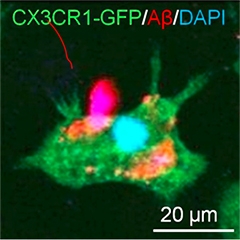
Dark Side of Microglia.
A newly identified type of microglia (dc) that engulfs Aβ (black) and expresses TREM2 appears dark in electron micrographs. [Courtesy of Marie-Ève Tremblay.]
“I got the sense from this meeting that people want to work more closely together,” said Beth Stevens, Children’s Hospital, Boston, who co-organized the microglial symposium together with Richard Ransohoff from Biogen, Cambridge, Massachusetts. Stevens emphasized the importance of collaboration for a field just beginning to get traction. “We now know what we need to do. We have a road map, the tools, and the insight. We can work individually in our own labs, or work together and make much more rapid progress,” she told Alzforum. Bradley Hyman, Massachusetts General Hospital, noted the value of having meetings on these two topics run side by side. Hyman co-organized the neurodegeneration symposium together with Adriano Aguzzi, University of Zurich, and Ricardo Dolmetsch from Novartis Institutes for BioMedical Research, also in Cambridge. “The cross-talk was really valuable, especially given how genetics now emphasizes that microglia and astrocytes are not secondary players but more primary drivers of disease,” Hyman said.
Some dogmas fell at this meeting even as researchers navigated new waters. Marco Prinz, University of Freiburg, Germany, put the kibosh on the prevailing idea that only brain-resident microglia originate from the yolk sac early in development. He reported evidence that seemed to convince even the most die-hard fans that non-parenchymal macrophages such as perivascular, meningeal, and choroid plexus macrophages are in fact long-lived macrophages that derive from yolk-sac progenitors, rather than bone-marrow-derived cells that turnover rapidly. “You can throw the text books out,” noted Gary Landreth, Stark Neurosciences Research Institute at the Indiana University School of Medicine in Indianapolis. “I see nothing wrong with that data set,” he told Alzforum. Ransohoff agreed. “I was completely shocked by that, but I’ll survive,” he quipped. “I think this is a robust finding, at least in mice, and we don’t have a lot of evidence that people are any different from mice in this respect.”
In general, scientists at the meeting agreed that they need to learn much more about microglial function in the normal brain before they can begin to understand how these cells influence disease. From potassium channels that regulate how microglial branches move and survey the brain, to active enhancers and transcriptome profiles that characterize these cells, researchers appear poised to more rigorously characterize microglia than just a few short years ago.
They are also developing better culture systems. A recurring theme at Keystone was that microglia de-differentiate rapidly—within eight hours—once they are taken out of their normal brain environment. While this knowledge left scientists questioning the relevance of most existing in vitro studies, they also agreed that it presents a valuable opportunity to understand what makes microglia tick. “If we can get these cultured cells back to looking like they do in vivo, we may have learned truly something about the brain,” noted Christopher Glass, University of California, San Diego, who gave the keynote address at the microglia meeting.
Along that vein, several groups reported that they had succeeded in creating microglia from human induced pluripotent stem cells—a first for the field. One group reported that when co-cultured with astrocytes and neurons, these induced microglia adopt features of brain microglia, including processes that extend and retract. “There goes another dogma,” noted Stevens. “If you had asked me a week ago if we could make induced microglia in vitro I’d have said, ‘No.’ Now three groups have reported on it.” Studying these cultures may bring the field closer to understanding microglial in the brain, researchers agreed.
Astrocytes got plenty of love at this meeting, too. For one, Shane Liddelow from Ben Barres’ lab at Stanford University, California, reported on different activation states of reactive astrocytes. To the chagrin of some researchers who have come to question the M1 and M2 designations for microglial activation, which many believe is too simplistic a nomenclature, Liddelow called these A1 and A2. He said the A1 astrocytes associate with neurotoxicity. Liddelow reported being able to culture these astrocytes in vitro and has identified several markers that characterize them, exciting other researchers who now plan to look for them in autopsy samples.
Researchers also reported stark differences between male and female microglia. This apparently is true both developmentally and later in life, prompting a re-evaluation of age- and sex-related risk factors for disease. On another curious note, Marie-Ève Tremblay from Laval University, Quebec, described a new type of microglia that show up as being particularly dense under the electron microscope. While their origin and function have yet to be determined, these “dark” microglia appear to occur in disease states and reside near blood vessels, close to amyloid plaques in mouse models of AD. They express TREM2, a microglial cell-surface receptor and now much-studied AD risk factor.
On TREM2 itself, some reported that this receptor may be involved in inflammation, and that it is important for microglial activation, proliferation, and phagocytosis. Scientists have long puzzled about what ligands might activate this receptor, and at Keystone, evidence emerged that it binds ApoE, ApoJ, and low-density lipoprotein. TREM2 also binds Aβ when the latter forms a complex with LDL, according to one presentation, whereupon microglia take it up the peptide and degrade it. TREM2 variants that associate with disease seem less effective in this respect.
On the neurodegeneration side of the Keystone hallways, researchers noted commonalities among various neurodegenerative diseases, from the biophysical structure of amyloid-forming proteins right up to how they scupper cells, both cell-autonomously and otherwise. Scientists also showed new data on how novel therapeutics—from dual leucine zipper kinase inhibitors to zinc-finger translational repressors that can be tailored to shut off transcription of specific genes—may work across a spectrum of diseases. Check back in the coming days as Alzforum brings you details on some of the highlights of the joint meeting.—Tom Fagan
Induced Microglia Make Debut at Keystone Symposium
Ironically, microglia in a dish stole some of the limelight at “Microglia in the Brain,” a symposium held jointly with another called “Common Mechanisms of Neurodegeneration,” June 12-16 in Keystone, Colorado. No sooner had Christopher Glass cautioned in his keynote address that isolated microglia bear little resemblance to their forbears in the CNS than posters and talks about induced microglial went up—and captured the imagination of meeting-goers. While time will tell how well this new type of cultured cell mimics microglia in their natural environment, initial signs are they that have a more authentic look and feel than microglial cells currently being used in vitro. Scientists seemed buoyed by the prospect of using the new cells for questions that cannot be addressed in studies of living animals or people. “These developments are very exciting,” said Irene Knuesel, Roche Innovation Center, Basel, Switzerland. “These may not be in vivo microglia, but they provide us with a tool and an opportunity to start studying these cells in much more detail.”
Three research groups independently generated induced microglia. One group cultured progenitors together with neurons and/or astrocytes; another used a “cocktail” approach, adding just the right combination of trophic factors to cause progenitors to morph into microglia; and the third combined both approaches, which they said provides more flexible experimental platforms.
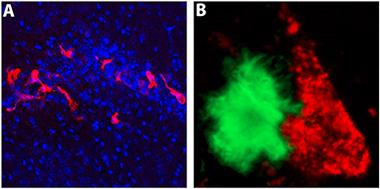
Hello microglia?
An iPSC-derived microglia-like macrophage (green) captures Aβ. [Courtesy of Florent Ginhoux and Kazuyuki Takata.]
Florent Ginhoux—known for his pioneering studies of the developmental origin of microglia—tried to recapitulate the in vivo development of microglia in an in vitro setting. In 2010, Ginhoux, who works at the Agency for Science, Technology and Research in Singapore, reported that brain-resident microglia derive from progenitors in the embryonic yolk sac (Ginhoux et al., 2010). “We basically wanted to recapitulate this primitive hematopoiesis in a dish,” Ginhoux told Alzforum. He developed a unique protocol that turns induced pluripotent stem cells (iPSCs) from healthy donors into yolk sac macrophages within a few weeks. Just like real macrophages, the cells are big and round; however, when Ginhoux cultures them together with neurons they develop microglial-like processes that reach out and touch the neurons. They also ingest synthetic Aβ fibrils (see image at left).
“It is fascinating that we can see very clear interactions between the two cell types,” said Ginhoux. While he agreed that it will be impossible to recapitulate the complexity of the brain, he plans to use iPSCs from healthy donors and people with neurodegenerative disorders, such as Parkinson’s and Alzheimer’s, to derive and co-culture neurons and glia and study interactions that may be important in disease.
Researchers working in Rudolph Jaenisch’s lab at the Whitehead Institute for Biomedical Research, Cambridge, Massachusetts, used a monoculture approach. Julien Muffat first derived primitive macrophages from human iPSCs, then used a fully defined culture medium that he developed to turn them into microglia. Muffat showed that these microglia become ramified, sporting processes with first-order branching, and that they phagocytose Aβ fibrils. Expression analysis suggests the cells’ transcriptome closely mimics that of fetal and adult microglia, rather than blood macrophages. He also said that his protocol was very robust and he has derived microglia from more than 20 different iPScell lines so far.
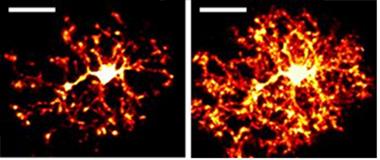
Cultured microglia.
Microglia derived from induced pluripotent stem cells extend cellular processes, just like microglia in the brain. [Courtesy of Julien Muffat, MIT.]
Muffat was able to shift that transcriptional signature closer to that seen in microglia in the brain by culturing the induced microglia in three dimensions with neurons and astrocytes. In the three-dimensional cultures, the microglia develop long processes that extend and retract (see image at left). In a laser-ablation assay, where researchers make a tiny wound by burning a hole through tissue, the induced microglia extended toward, and clustered around, sites of injury, much like microglia do around amyloid plaques in the brain. In cerebral organoids the induced microglia cells appear to “tile” the way they do in vivo in the brain.
Muffat is now collaborating with researchers in Rudy Tanzi’s lab at Massachusetts General Hospital, Charlestown, to embed these cells in three-dimensional neuronal cultures that generate amyloid plaques and neurofibrillary tangles, the hallmarks of AD (see Oct 2014 news).
Also taking a more direct stab at developing induced microglia, Edsel Abud in MathewBlurton-Jones’ lab at the University of California, Irvine, shied away from using neurons or other cells. “The main difference with our approach is that it allows us to study microglia without the need for complex co-culture systems,” said Blurton-Jones. “One potential advantage is that it will be easier to conduct high-throughput quantitative screening.” Some pharma researchers at the meeting lamented that they cannot carry out high-throughput screens on microglia using tissue slice preparations, but thought this approach might prove valuable for translational research.
Abud built on years of studies on mouse microglial ontology that highlight not only where microglia originate, but also what they need to thrive. He told Alzforum that he tried to identify from the literature the components that seemed necessary for microglia to survive, mature, and stay healthy, and then tested those components empirically. “We thought we’d try a reductionist approach and find the soluble factors that would be needed to derive and maintain microglia,” he said. Abud modified a protocol used to derive primitive hematopoietic progenitors from iPS lines, and then guided those cells toward a microglia-like phenotype with a mixture of different factors.
iPS-microglia in the brain.
When transplanted into the hippocampi of immune-deficient 5xfAD mice, microglia derived from induced pluripotent stem cells (red) migrate and interact with Aβ plaques (green). [Courtesy of Matt Blurton-Jones, UC Irvine.]
These iPS microglia (iPS-MG) express typical microglial markers, including P2Y12R, TREM2, DAP12, and CX3CR1. Based on co-expression of P2Y12R and TREM2, these cells are more than 96 percent pure, said Blurton-Jones. Abud and colleagues used three-dimensional principle component analysis, a method to pull out the most significant trends in a data set, to compare whole-genome transcriptional profiles of the iPS-MG against those of other cells. The iPS-MG profiles matched closely with those of adult and fetal microglia but diverged from those of blood monocytes.
Blurton-Jones and colleagues are studying how the iPS-MG respond to Aβ plaques and neurofibrillary tangles, the hallmarks of AD pathology. On his Keystone poster, Abud reported that the cells phagocytose fibrillar Aβ, and upregulate several AD-associated genes in the process, including TREM2, ApoE, ABCA7, and PICALM. Clusterin stood out for its 35-fold elevation in expression. In collaboration with Rakez Kayed at University of Texas Medical Branch, Galveston, Abud found that the cells also take up tau oligomers isolated from human brain. Working with Karen Gylys from the University of California, Los Angeles, the group also found that iPS-MG could phagocytose human brain-derived synaptosomes and that blocking components of complement receptor 3 (CR3) could impair this process. Researchers led by Beth Stevens at Children’s Hospital, Boston, have shown the microglia prune synapses in the adult brain in a CR3-dependent manner, and that this can be exacerbated by Aβ (see Apr 2016 news).
Ginhoux noted that having these different ways to make induced microglia is good for the field. “This is how we progress in science. It should give others the opportunity to build and improve on these approaches,” he said.
Keynote speaker Glass, of the University of California, San Diego, said that going from iPS or embryonic stem cell to in vitro microglia represents a huge advance for the field. “It seems that these researchers have made substantial progress,” he told Alzforum. But he cautioned that in vitro cells may not always tell us what happens in vivo. On that score, Blurton-Jones and colleagues have transplanted induced microglia into the hippocampus of immune-deficient 5xFAD mice (Marsh et al., 2016), where they began to migrate toward and interact with Aβ plaques (see image above). Glass also emphasized the need to study human microglia in a human brain context, and suggested organoids might be the way to go. Muffat and colleagues are partway there with their three-dimensional culture systems.
All three groups plan to study how genetic variation affects their induced cells. “We can now try to figure out how variants in genes such as TREM2 affect the different cell types in the brain,” said Ginhoux. Blurton-Jones and colleagues have already begun to culture induced microglia from people carrying the R47H and T66M TREM2 mutations, which are risk alleles for Alzheimer’s and frontotemporal dementia, respectively.
The researchers agreed that these cell lines can help the field get past a niggling problem in studying risk alleles identified by genome-wide association studies. As emphasized by Glass and others at the meeting, genetic heterogeneity in the population presents a major hurdle in interpreting the effect of GWAS hits on disease pathology. To model this, Ginhoux, Muffat, and Blurton-Jones plan to use CRISPR technology to introduce genetic variants into stable microglial lines. In Keystone, Muffat reported using CRISPR to generate induced microglial with the presenilin 1 A246E mutation and with TREM2 knocked out. “Using isogenic lines will help us account for background genetic variation that might confound analysis,” noted Blurton-Jones.—Tom Fagan
Long given short shrift by neuroscientists, microglia are blossoming into darling research subjects. The newfound attention comes courtesy of genetic evidence implicating microglia in neurodegenerative diseases and cellular evidence for a role in pruning synapses. While science still knows little about what makes these immune cells tick, that’s beginning to change. At “Microglia in the Brain,” a joint symposium held June 12-16 together with “Common Mechanisms of Neurodegeneration,” in Keystone, Colorado, researchers revealed a potassium channel (THIK-1) that drives microglia to survey their milieu.
In the healthy brain, microglia constantly extend and retract finger-like projections, presumably to feel around for signs of trouble such as foreign invaders or dying neurons, and to sense the functional state of synapses. Repeated scanning of microglia in the brain show that they can probe the entire organ within a few hours. What drives this behavior? Scientists at Keystone agreed that answering this question is fundamental to understanding how microglia function in the brain. Taking a stab at the problem, David Attwell, University College London, reported that an outwardly rectifying potassium channel drives microglial surveillance.
Feeling About.
Images of a mouse hippocampal microglia captured at a single time point (left), and cumulatively over 20 minutes (right), show how it surveys its environment. [Image courtesy of David Attwell and Christian Madry.]
Attwell and collaborators identified the potassium channel by studying cell motility in mouse brain tissue slices. They began by probing responses in the classic laser-injury model, whereby microglia flood in to repair a hole burned by a laser. In this paradigm, two things happen in microglia, he said. Cytosolic calcium levels rise, and potassium exits the cells via an ion channel. However, the responses are independent of each other, said Attwell. He knows this because thapsigargin, which blocks the mobilization of calcium from internal endoplasmic reticulum stores, has no effect on the potassium efflux.
Adding ATP to microglia evokes both these responses, hence they appear to be initiated by purinergic receptor signaling. Microglia abundantly express several of these receptors, which are activated by ATP released by dead and dying cells. Scientists believe that ATP and other purines spur microglia into action to tidy up cellular debris. Attwell showed the P2Y12 purinergic receptors primarily trigger the potassium current. PSB 0739, a P2Y12R antagonist, suppressed it, while blocking other purinergic receptors had no effect on potassium efflux. “This is phenomenally important work,” noted Richard Ransohoff from Biogen in Cambridge, Massachusetts. “It gives us a molecular explanation [for motility] and therefore numerous genetic and pharmacological tools to manipulate it,” he told Alzforum.
Channeling Ions.
Purinergic receptors may trigger release of potassium in microglia. [Image courtesy of David Attwell.]
While the P2Y12 receptor may trigger the potassium release, which ion channel conducts it? Attwell found that blocking voltage-dependent K+ channels had no effect. This left inward rectifying and two-pore domain K+ channels as the only other choices, he said. The electrophysiology of the former does not match that of the current driven by the P12YR, hence Attwell concluded that he was chasing a two-pore domain channel. Analysis of published transcriptomes suggested microglia express only one gene of this kind, i.e., that encoding the two-pore domain halothane-inhibited K+ channel, aka THIK-1.
This channel sits in the cell membrane. It is widely expressed in mammalian tissue, including the brain where it is found mainly in microglial cells. It shares only 25-35 percent identify with other two-pore K+ channels. These channels have not been well-studied, but their regulation seems complex.
What might THIK-1 do in microglia? Ironically, Attwell found that it is not required for targeted motility; in other words, the cells still flood in to repair damaged tissue when that channel is blocked. But resting surveillance was another story. Blocking THIK-1 dramatically reduced extension and retraction of microglial projections in tissue slices, as well as the ground they managed to cover. The same occurred in vivo, said Attwell. Furthermore, the THIK-1 channel seems to be required for a key immune response of microglia, that is, their release of certain cytokines. Blocking THIK-1 prevented secretion of interleukin 1β in response to inflammatory signals.
Researchers at the meeting were both impressed by the data and concerned about its implications. This channel, and hence microglial baseline activity and immune response, can be blocked by gaseous anesthetics such as halothane and isoflurane, which are often used in research studies. “It’s hard to overstate how crucial that is,” noted Ransohoff. Researchers were concerned that microglial responses may be muted under anesthesia. “When we do experiments on live mice, particularly two-photon imaging, what can we use for anesthesia?” Ransohoff wondered.
Attwell reinforced another important theme that emerged at the meeting, namely that isolated microglia in vitro poorly mimic their brethren in the brain (see related Keystone news). Cultured microglia express little or no THIK-1, though perhaps they compensate with a different potassium channel or regulate motility in a different way, he suggested.—Tom Fagan
People often associate Schrödinger with the observer effect, namely that you cannot measure something without perturbing it. Are researchers studying microglia facing an observer paradox of their own? Just how fickle these immune cells truly are dominated conversations at “Microglia in the Brain,” a Keystone symposium held June 12-16 in Keystone, Colorado. Recently researchers have begun using transcriptomics and genomics to characterize these cells from human and mouse brain and figure out how they respond to environmental cues. A take-home message from this symposium was that once yanked out of their normal environs, microglia can change so radically that they no longer appear to be microglia.
In his keynote address, Christopher Glass, University of California, San Diego, emphasized the de-differentiation that microglia undergo. Once you remove microglia from the brain, Glass said, their transcriptome changes fast and the transcripts that fade the most are those that made the cells unique in the first place. He reported that the expression of microglial-specific genes drops fourfold within six hours of culturing the cells in vitro. The change is usually complete by eight hours. “That’s on the same time scale as how long they take to sense their local environment in vivo,” said Glass, suggesting that microglia need constant signals from their surroundings to maintain their phenotype.
Glass attributed these expression changes to a dramatic reorganization of gene enhancers. His group had previously reported that the local environment influences the complement of active enhancers that drive gene expression in microglia and macrophages (see Feb 2015 conference news on Gosselin et al., 2014). Enhancers are regions of DNA that bind transcription factors determining cell lineage, and they are subject to epigenetic regulation. Any given cell contains 20,000 to 30,000 active enhancers, of which up to 300 are deemed “super enhancers.” “These are DNA regions where the cell has collected a lot of resources to regulate expression, so we think they are particularly important,” Glass said. The function of about 25 of the microglial super enhancers is known; they regulate expression of genes such as the fractalkine receptor and the microglial transcription factor PU.1. “We don’t know much about the other genes covered by these super enhancers,” said Glass.
Glass noted that when microglia are removed from their natural habitat, half of the super enhancers fade away. The significance of this genetic reorganization worried researchers at Keystone, given that many studies use isolated microglial cell lines. Researchers seemed to agree that those cells may be useful in addressing certain questions, but that they poorly reflect the overall behavior of microglia in vivo. To come to grips with this, researchers have begun to profile the transcriptomes of microglia from the brain to determine what makes these cells unique and how they respond to different environmental challenges, such as inflammation or Aβ pathology.
Researchers from Joe El Khoury’s lab at Massachusetts General Hospital proposed a mouse microglia “sensome.” This would be a cadre of transcripts encoding proteins and receptors involved in detecting ligands and microbes (see Mar 2013 conference news and Hickman et al., 2013). Oleg Butovsky and colleagues at Brigham and Women’s Hospital, Boston, identified a purely microglial transcriptome that depends on TGF-β signaling (see Butovsky et al., 2014). Other groups have analyzed microglial transcriptomes in various mouse models of disease, such as neurodegeneration or glioblastomas.
Signature Transcriptomes
How do these transcriptomes compare, and what can they tell us about pathology? Brad Friedman, a computational biologist at Genentech, South San Francisco, addressed this by analyzing 18 different data sets looking for microglial genes that are co-regulated, i.e., that tend to be perturbed in the same way. In Keystone, Friedman reported on large clusters of genes that are uniquely regulated in different disease models, perhaps reflecting distinct ways in which microglia are activated. For example, he found one cluster that responds to interferon and another that drives cell proliferation. He also found a “neurodegenerative disease cluster” of about 100 genes that are common to PS2APP and 5xFAD models of AD, and to an SOD1 model of ALS. This cluster showed no response to infection or when mice are challenged with lipopolysaccharide, which elicits robust immune responses in mice. The implication is that this cluster reflects a unique neurodegenerative signal, but whether it represents a toxic or neuroprotective response is not known. Friedman wants to study this and also what makes this signature turn on and off. “If we could modulate it, perhaps we could alter the course of disease,” he said. The neurodegenerative disease expression signature seemed to increase in older mice compared to younger, and in the cerebellum over other brain regions.
Intrigued by these signatures, scientists at the meeting peppered Friedman with questions. Has he looked at tau models? (He has not.) Are there disease-specific signatures? (He had no specific examples.) Could he identify some curiously unlabeled clusters on his slides? When pressed, Friedman revealed that one of them was from a knockout mouse that might model AD, but this cluster poorly overlapped with the neurodegenerative disease cluster.
While Friedman didn’t elaborate on individual genes in this neurodegenerative cluster in his talk, he told Alzforum that ApoE was one of them. “I found that interesting because people think ApoE is mostly expressed in astrocytes,” he said. The cluster was highly enriched in transcripts for lipoprotein receptors and transmembrane and secreted proteins, said Friedman. This suggests the microglia may be engaging more with their environment. IGF-1, thought to be neuroprotective in ALS, turned up in the cluster as well.
Some researchers queried Friedman about batch effects. How valid is it to compare expression levels across different data sets? Friedman said he used z-score normalization, a statistical method that allowed him to compare relative expression levels across different studies, with each study serving as its own control.
Whether these mouse transcriptome signatures reflect what goes on in the human brain remains to be seen. Researchers are only just beginning to profile microglia from human tissue, and they face many hurdles, not least being access to fresh tissue. Using a dataset generated at Genentech of gene expression in the fusiform gyrus of people with Alzheimer’s disease, Friedman looked to see if the microglial neurodegenerative signature turns up. The researchers had chosen this area of the brain because samples are easier to obtain than the more highly sought-after hippocampal specimens and the gyrus is also one of the earliest affected regions in the disease. While acknowledging that the data are from bulk tissue, and so include all cell types, Friedman reported that two-thirds of the 100 genes in the mouse neurodegenerative disease signature were differentially expressed in the AD samples, as well.
Several other groups are trying to analyze purified microglia from human brain. Erik Boddeke, from the University of Groningen, The Netherlands, takes fresh postmortem samples from the Dutch brain bank and also through a collaboration with the University of São Paulo, Brazil. “The major bottleneck in the field is ready availability of sufficient postmortem tissue samples,” Boddeke told Alzforum. Because he wants to examine healthy tissue and extract sufficient numbers of viable microglia for study, only about a quarter of the samples he obtains turn out to be suitable.
Even so, Boddeke has made some progress comparing transcriptome profiles of human and mouse microglia. While many genes are similarly expressed, he said there are also substantial numbers of human microglia-specific genes that turn out to be not specific to microglia in rodents. Researchers at Keystone thought that was troubling for a field that relies so heavily on mouse data.
Boddeke further reported that aging changes mouse and human microglial transcriptomes in different ways. Microglia from older mice ramp up expression of genes involved in inflammatory responses, whereas the older human microglia lean toward genes involved in neuronal support. Boddeke found that microglia from mouse models of AD and ALS express aging-like inflammatory genes. Glass considered Boddeke’s study very interesting. Working with Fred Gage at The Salk Institute, La Jolla, California, Glass studies biopsy samples taken from children who have brain tumors or epilepsy. While he cautioned that it is extremely challenging to obtain viable cells from postmortem tissue, he noted that Boddeke’s core transcriptome looked like it might turn out to be very similar to the one he identified in the samples from children. “Given that the demographics of the samples are completely different, the similarities in the data may be pointing to genes that are most highly expressed in microglia and that make them most different from other macrophages,” Glass said.
Going forward, Boddeke plans to compare transcriptomes of microglia from healthy people with those at various stages of AD. He admits the major problem will be getting sufficient numbers of high-quality, carefully staged samples. Even so, the transcriptome analysis may identify new microglial markers, since some of the genes in the microglial-specific transcriptome encode cell surface proteins.
Glass uses human microglial transcriptomes to assess a different problem, namely heterogeneity in gene expression between one person and the next. This, he said, presents a challenge for the field, but also an opportunity. Glass told Alzforum that so far, he has studied samples from about 20 different children, and while the expression levels of some microglial genes are similar, others vary considerably. For example, Glass noted a fourfold difference in TREM2 and dramatic differences in ApoE expression. “It’s possible we might be able to predict such quantitative changes based on genotype,” said Glass. “That could ultimately help us predict what is going on in the brain in response to environmental stress.”
Heterogeneity in microglial gene expression could make it difficult for researchers to interpret the effects of hits from AD genome-wide association studies (GWAS). Many variants uncovered by those studies lie in non-coding regions in microglial genes and are thought to weakly influence gene activity. How to distinguish these subtle effects against background noise? Here again, Glass thinks expression analyses could help. The key point, he said, is that regulatory regions in microglia are very different from regulatory regions in neurons or astrocytes, therefore a cell-specific approach is needed to understand the effect of a given genetic variant on these cells. “If we can overlay microglial enhancer atlases on GWAS loci, then we may obtain very useful information.”
It became clear at the meeting that scientists need to work out how to keep track of the reams of transcriptome data that are beginning to flow from different labs. Boddeke has set up a microglial gene expression database (see Holtman et al., 2015) where researchers can deposit and access transcriptomics data. While some individual labs maintain their own databases, Boddeke said it will be immensely valuable for the field to have access to data in a standard format that is as open as possible. He plans to take an expanded version live in October.—Tom Fagan
Unbiased Screen Fingers TREM2 Ligands That Promote Aβ Uptake
Since scientists found that variants in the gene for TREM2 triple a person’s risk for Alzheimer’s, they have wondered what the protein does in health and disease. Talks and posters at “Microglia in the Brain,” the Keystone symposium that ran June 12-16 in Colorado, addressed some of the main issues, including what ligands bind the cell surface receptor and whether it helps or hinders Aβ pathology in mouse models of AD.
Binding Array.
From a large library of extracellular and membrane proteins, ApoA1, LDL, and malondialdehyde-modified LDL bound TREM2. [Image courtesy of Felix Yeh and Neuron.]
The TREM2 gene codes for a single transmembrane protein that partners with the adapter protein DAP12. Scientists believe that the two proteins trigger a series of downstream signaling events, but what kicks them into gear remains a bit of a mystery since few ligands for TREM2 have been identified. Felix Yeh, working with Morgan Sheng, Lino Gonzalez, and colleagues at Genentech, South San Francisco, decided to tackle this issue by using an unbiased screen. Yeh used a protein microarray developed at Genentech to study the interactions of extracellular proteins. The array contains thousands of secreted and transmembrane proteins that can be tested against proteins of choice. When Yeh probed it with the extracellular domain of TREM2, it bound several lipoproteins, including apolipoprotein A1 (ApoA1), low-density lipoprotein (LDL), and unc-5 homology B (see image at left). Using a biophysical technique called bio-layer interferometry, which directly measures protein-protein interactions, he confirmed that LDL and ApoA1 bind TREM2. UNC5B failed to bind in this assay.
This made Yeh wonder if TREM2 might bind apolipoproteins, including ApoE and ApoJ, aka clusterin, that can alter the risk for AD. Sure enough, in the interferometry assay, he found that both, when in lipidated form, bound the receptor. Yeh said his initial screen likely missed these proteins because they are not lipidated on the microarray.
Does cellular TREM2 bind these proteins? Yeh and colleagues tested this in HEK293 transfected with a doxycycline-driven TREM2 gene. When he induced expression of the surface receptor, the cells took up acetylated LDL (acetylation of the protein prevents its binding to the LDL receptor, which is also expressed on HEK cells and might interfere with the uptake assay). The TREM2-expressing HEK cells also took up lipidated ApoE and lipidated clusterin. Turning to microglia, the researchers found that cells from TREM2 knockout mice took up LDL much more slowly than did cells from wild-type mice, supporting the idea that TREM2 plays a role in microglial lipoprotein uptake.
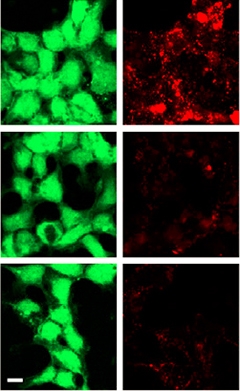
Lipoprotein Uptake.
HEK293 cells expressing wild-type TREM2 (top) readily ingest LDL (right); cells expressing Y38C (middle) and R47H (bottom) variants of TREM2 do not. [Image courtesy of Felix Yeh and Neuron.]
To relate these observations to disease, the researchers tested a variety of TREM2 mutations in the protein-binding and cell-uptake assays. They found that the Y38C and T66M mutations, which cause Nasu-Hakola disease and have been linked to frontotemporal dementia, abolished TREM2 binding to LDL, clusterin, and ApoE. The AD risk alleles R62H, D87N, and R47H, in that order, progressively weakened binding, such that R47H TREM2 bound about half as much lipoprotein as did wild-type receptor. Uptake assays told a slightly different story, perhaps because of the presence of other receptors in the cells and different sensitivities of the assays, said Yeh. HEK cells expressing Y38C and T66M TREM2 took up about a fifth as much acetylated LDL (see image at right), which gibes with their reduced binding. Even though R47H TREM2 binds more tightly to the lipoproteins, cells expressing this mutant took up similar amounts of LDL to the Y38C and T66M cells. R47H cells did take up about half as much clusterin as did wild-type cells, though. Finally, cells expressing the R62H and D87N variants, which have the highest affinity for lipoprotein among the tested mutants, took up nearly normal amounts of clusterin but slightly less LDL.
Lastly, Yeh tested if these perturbations in binding and uptake of apolipoproteins weakened microglial processing of Aβ. He reported that wild-type, TREM2+/- heterozygotes, and TREM2 knockout cells all took up very little free Aβ. In contrast, all cells imbibed vastly more Aβ when it came combined with LDL or clusterin. However, there was a marked TREM2 dose-dependency—homozygotes took up about twice as much as did knockouts, with the heterozygotes falling in between. Since Aβ binds ApoE and clusterin, and both proteins are found not only in the extracellular space but in amyloid plaques, these findings raise the possibility that microglia are particularly suited to digesting plaque-associated Aβ, suggested Yeh.
Researchers at Keystone were impressed by the findings, which were published in the July 20 Neuron, and Yeh’s poster drew a large crowd. “There have been some attempts to find TREM2 ligands, but this stands out for the rigor of the analysis,” said Gary Landreth, Stark Neurosciences Research Institute, Indiana University School of Medicine, Indianapolis. Using a candidate approach others had found that ApoE binds TREM2 and that the R47H mutation weakens the interaction, but they had not made the connection to Aβ uptake (see Atagi et al., 2015; Bailey et al., 2015).
Yeh’s findings may be directly relevant to human biology. He said that among blood samples donated by 1,500 volunteers, he found one R62H carrier whose macrophages took up less lipoprotein-Aβ than did macrophages isolated from an age-, sex-, and ethnicity-matched control.
What about other microglial responses that TREM2 mutations might perturb? In her poster, Fargol Mazaheri, who works in Christian Haass’ lab at the German Center for Neurodegenerative Diseases, Munich, described a transcriptomics approach to nail down what knocking out TREM2 does to a cell. She isolated microglia from normal mice and from TREM2 knockouts with a fluorescent-activated cell sorter. Then she quantified transcripts using nanostring technology, à la Oleg Butovsky’s seminal study (see Butovsky et al., 2014).
Mazaheri’s data suggest that TREM2 functions in microglial homeostasis, migration, and inflammatory responses. Among 122 genes thought important for homeostasis, many were upregulated in TREM2 KOs, while genes involved in inflammation and migration were turned down. Mazaheri tested cell motility by juxtaposing old and young organotypic brain tissue slices together in culture. In this scenario, CD68-positive cells (microglia, macrophages, and monocytes) usually migrate from the young tissue toward the old, she explained to Alzforum, perhaps to scavenge dead or dying cells. Not in the case of TREM2 KO tissue. To test mobility specifically in microglia, Mazaheri took advantage of CRISPR technology to knock TREM2 out of N9 microglial cells. This repressed mobility-related genes and chemotaxis. In preliminary experiments she tested microglial migration in vivo in mice using a laser injury model, in which microglia flock to the site of a small wound burned into tissue. Mazaheri found that TREM2 knockout microglial are sluggish in this assay. The findings dovetail with reports that in TREM2-deficient AD models, microglia don’t surround plaques as readily as they normally do (see Jun 2014 news and May 2016 news).
Researchers at the labs of Landreth and Bruce Lamb, now also at Stark, have also used CRISPR/CAS9 to study TREM2 biology. They generated R47H TREM2 heterozygotes and crossed them to APPPS1 mice. Preliminary data point to fewer microglia and reduced microglial proliferation in these crosses. The R47H TREM2/APPPS1 mice also have fewer plaques at four months of age than APP controls. This fits with prior reports from these labs that APP/PS1 mice lacking TREM2 had fewer plaques in the hippocampus than controls (see Dec 2014 conference news and Jay et al., 2015).
Those earlier reports surprised the field, which had come to expect that TREM2 might play a role in microglial plaque clearance. The data also contrasted with results from Marco Colonna at Washington University, St. Louis, who found more hippocampal plaques in 5xFAD mice lacking TREM2 (see Feb 2015 news and Feb 2015 conference coverage). Researchers were left wondering whether TREM2 promotes clearance or accumulation of plaques.
In his talk, Landreth seemed to settle this controversy. Updating his characterization of the APP/PS1 TREM2 knockouts, he said his lab’s data now better matches that of WashU group. “We are now on the same page with Colonna and colleagues,” he told Alzforum. The reason? Aging. Taylor Jay, a graduate student working for both Landreth and Lamb, had initially necropsied four-month-old mice, but when she examined eight-month-old animals, she saw that the TREM2 knockouts had accumulated more plaques in the hippocampus than the regular APP/PS1 mice, just as Colonna’s group had seen in their eight-month-old mice. “We see a distinct progression-dependent change in plaque burden” said Landreth. Researchers at Keystone wondered what might underlie that change, but Landreth was not sure. He suspects a dynamic up-down regulation of plaque accumulation related to TREM2 function, but what drives that dynamic is unclear, he said.
Further analysis will tell if plaque burden changes as the R47H TREM2/APPPS1 animals age as well.—Tom Fagan





Comments
No Available Comments
Make a Comment
To make a comment you must login or register.