The fields of neuroscience and immunology melded into one at “Neuroinflammation in Diseases of the Central Nervous System,” a Keystone Symposium held January 25-30 in Taos, New Mexico. The behavior of myeloid cells in the brain drew close scrutiny at the conference, as researchers grappled with fundamental questions such as, what do microglia actually do, and how do they respond to and influence the course of neurodegenerative disease? Novel genomic and proteomic tools emerged that may help answer these questions. Researchers abandoned the old M1/M2 microglial phenotypes calling for more relevant characterizations, and embraced TREM2 and other molecules that play central roles in microglial responses. Read Jessica Shugart’s stories on the meeting’s highlights.
Neuroinflammation Field Grapples With Complexity at Keystone Symposia
Ponder, for a moment, the opening slide for the Keystone symposium “Neuroinflammation in Diseases of the Central Nervous System,” which ran January 25-30 in Taos, New Mexico. It defined neuroinflammation as “inflammation of a nerve or parts of the nervous system.” “When you need to use part of the word you’re defining to define it, then no one really knows what the word means,” joked Richard Ransohoff. The uncertainty epitomizes the field, which despite many years of research may only now be finding its groove. Three hundred attendees ascended to the high desert to share data and more thoroughly define and understand neuroinflammation. Co-organized by Ransohoff, who recently moved from the Cleveland Clinic in Ohio to Biogen IDEC in Cambridge, Massachusetts, Chris Glass of the University of California, San Diego, and V. Hugh Perry of the University of Southampton in England, this was the first Keystone symposium dedicated to neuroinflammation since 2005, when only 164 articles popped up on PubMed related to the topic. Last year, that number was 10 times higher, and the meeting pulsed with the energy of a rapidly changing field. Stay tuned in the coming days as Alzforum brings you findings and controversies that engaged scientists at the meeting.
Sangre de Cristo Mountains.
New Mexico, an apt location to debate the vast field of neuroinflammation. [Image courtesy of Dave Hensley http://bit.ly/1FfWmW8.]
Amidst 27 oral presentations, 183 posters, and in conversation during meals, coffee breaks, and ski-lift rides, researchers grappled with how inflammation slows or hastens CNS disorders ranging from Alzheimer’s disease to multiple sclerosis, traumatic brain injury, viral infection, and stroke. Microglia and their monocyte cousins stole the show, as researchers aimed to more clearly understand what the brain’s in-house immune cells do, how they spring into action (or clam up) in the face of disease, and how they differ in form and function from infiltrating macrophages from other parts of the body.
Hunkered down at the Sagebrush Inn, once the home of the artist Georgia O'Keeffe, researchers painted a picture of complexity, sinking any hope of black-and-white explanations into the sienna desert mud. First and foremost, attendees tried to figure out what distinguishes microglia in the brain from other myeloid cells in the body. While circulating monocytes originate from progenitors in the bone marrow throughout a person’s lifetime, microglia derive from precursors that migrate from the yolk sac to the brain early in development. Once in the brain, these myeloid cells develop personalities uniquely suited to their new abode. As reported at the meeting, researchers uncovered combinations of transcription factors that buddy up and bind side-by-side to “super enhancer” regions in the genome to facilitate this specialization. These transcription factors were triggered by cues specific to each cell’s environment, and researchers reported that transferring macrophages into tissue culture dishes could to some extent undo their unique identities. How these signals might alter microglia in the aging or diseased brain was a hot topic at the meeting. Similarly, researchers reported that the gene expression profile of microglia and other macrophages changed dramatically in the context of diseases such as AD, ALS, and MS. While researchers made headway on distinguishing microglia from other macrophages at the level of gene expression, several attendees said it will be important to translate these genetic findings into functional ones.
Mixed Messages.
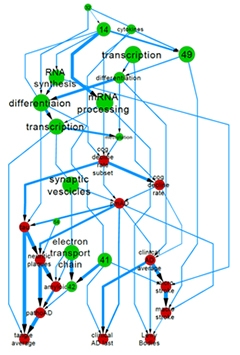
Constructed from genomic, clinical, and pathological data, this hierarchical network portrays causal relationships between groups of co-expressed genes and Alzheimer’s disease traits. Thicker arrows denote stronger relationships. [Image courtesy of Chris Gaiteri, Rush University, Chicago.]
At Keystone, researchers agreed it was time to bury the binary M1/M2 classification of pro-inflammatory and anti-inflammatory macrophages. There is consensus that, in vivo, pure M1 or M2 phenotypes, defined by the expression or lack of expression of specific genes, do not exist. Susanna Rosi of the University of California, San Francisco, reported that peripheral macrophages infiltrating the CNS after a traumatic brain injury, for example, express a variety of cytokines and do not adhere to strict M1 or M2 phenotypes. Christine Hsieh, also from UCSF, found no M1 or M2 profile in single cells extracted from the injury site following TBI. Perry received a round of applause when he said that adhering to the M1/M2 framework has held the field back, and that reviewers should no longer require researchers to consider this dichotomy. In many ways, the complexity of macrophage responses makes perfect biological sense, Perry told Alzforum. “You want diversity of function to respond to a diversity of insults. The last thing you want is for macrophages to behave in only one or two ways.” The tricky part, he added, is figuring out how to understand these complex behaviors in a coherent way.
Data about macrophage populations dovetailed with research on TREM2, clearly the most contentious molecule at the meeting. Some researchers reported that this cell surface receptor and AD risk factor predominantly shows up on microglia, while others claimed that infiltrating macrophages express most of the TREM2 in the brain. Researchers disagreed on TREM2's role in the context of AD—some reported that plaque burden fell in TREM2-deficient mice, others that plaques stayed the same, and still others that plaques increased. TREM2 emerged as a potential survival signal for microglia as they fight off plaques, suggesting its loss could kill microglia and limit plaque clearance. A new idea emerged that lipids activate TREM2; this piqued widespread interest. It’s unclear whether this happens in the brain, and which lipids could do the trick; researchers speculated about apoptotic cells or even lipidated ApoE complexed with Aβ.
Researchers at the meeting also reported that microglia do more than just clean up debris; they play crucial roles in synaptic maintenance. Microglia are known to prune synapses during development, but recent findings suggest they do the same in the adult brain. At Keystone, researchers reported that complement proteins trigger microglia to target weaker synapses and that early synapse loss in AD mouse models depends on the complement system. Aβ oligomers switched this pruning into high gear. Intriguingly, a “don’t eat me” signal seems to protect some synapses, and researchers are studying whether this signal could become jumbled in neurodegenerative disease. PirB, a receptor for MHC I that is also activated by Aβ, keeps synaptic plasticity in check, and inhibiting it could be a way to boost flagging synapses.
The perennial chicken-or-egg question came up repeatedly: Is neuroinflammation a cause or consequence of neurodegenerative disease? At the meeting, researchers attempted to place neuroinflammation within a hierarchical framework with other pathologies. Drawing from a large cohort of AD patients, Chris Gaiteri of Rush University in Chicago combined gene-expression profiles with clinical data to create a network model of AD. He identified hubs represented by groups of genes with a common function (such as synaptic regulation), or by pathological characteristics (such as Aβ deposition). He then created a hierarchy based on the hubs that most affected other components of the network. Cytokines appeared near the top of the chain, followed by synaptic genes and cognitive decline, with tau and amyloid pathology skirting the bottom. While the prominence of neuroinflammatory hubs and the relative obscurity of Aβ and tau surprised some researchers, others felt that these complex network approaches were the way forward. “That’s where we’re going to define targets for intervention, and find insight into how systems work,” said Perry.
Perry’s talk moved the discussion below the neck, focusing on the importance of systemic inflammation as a driver of neurodegenerative disease. The neurodegenerative process primes microglia for action, and inflammatory signals from elsewhere in the body could kick the cells into high gear and exacerbate disease, he said. This relationship may prove important in the context of aging, where inflammatory diseases such as obesity and atherosclerosis could contribute to the problem. Perry presented results from a small trial that suggested treatment of AD patients with etanercept, a systemic inhibitor of TNF-α, may have slowed cognitive decline.
Genomic stability in neurons may also crumble with aging and incite chronic inflammation, suggested Mark Albers of Massachusetts General Hospital in Boston. He reported that double-stranded RNA (dsRNA), a product of botched DNA repair, multiply as neurons age and can trigger an inflammatory response. Albers stumbled upon the idea when he discovered that some APP transgenic mice developed neurodegeneration while others did not. In the former, copies of the transgene had inserted in opposite orientations, so they were transcribed in both sense and antisense directions, resulting in dsRNA. This duplex triggered a receptor called MDA5, an intracellular pattern-recognition receptor evolved to recognize viruses, and raised an inflammatory alarm. Albers proposed that this response—which can be triggered by any dsRNA—damages neurons. He pointed out that dsRNA commonly forms as a result of genetic inversions that occur during the DNA repair process in aging neurons. Glass was intrigued by the talk, but said that since this process would occur independently in each neuron, single-cell genomic sequencing would be the only way to measure it. He added that Albers had been selected to give a talk because his data was the most “out there.” After hearing the talk, Glass said it still was.—Jessica Shugart
Nature Versus Nurture: What Gives Microglia Their Identity?
Microglia, the macrophages of the brain, took center stage at “Neuroinflammation in Diseases of the Central Nervous System,” a Keystone symposium held January 25-30 in Taos, New Mexico. Even though the spotlight has been shining on the shape-shifting glial cells for years, researchers are only now developing the tools to understand what they do in the normal brain, how they differ from macrophages that come from other parts of the body, and how they react to and influence the course of neurodegenerative disease. At the meeting, researchers presented a trove of findings that painted microglia as cells tailor-made for their brainy environment, yet quite capable of transformation—good and bad—when put under pressure. Part 2 of this Keystone series addresses microglial origins and how their local environment helps seal their fate. Part 3 will describe how the cells change in the diseased brain.
Day in and day out, multitudes of macrophages live in drastically different environments throughout the body. Some, such as microglia in the brain or Kupffer cells in the liver, stay put in their home organs throughout life, whereas others, such as blood monocytes, are nomads, primed to invade any tissue on a whim. Early in development, progenitors of microglia and other tissue-resident macrophages migrate to their destination and continue to supply those organs with macrophages throughout life (see Gomez Perdiguero et al., 2013). This is in stark contrast to blood- borne monocytes, which are continuously being replaced by hematopoietic stem cells in the bone marrow. In recent years scientists discovered that brain-resident microglia derive from cells in the embryonic yolk sac, but at Keystone Frederic Geissmann of King’s College London claimed those progenitors make a pit stop on the way.
Enhanced Assimilation.
Macrophages receive signals from unique proteins in their environments, which activate transcription factors that hook up with others to shape the macrophages’ behavior. [Image courtesy of Gosselin et al., Cell, 2014.]
Using an arsenal of transgenic mice engineered to express fluorescent markers at pivotal times during development, Geissmann and colleagues found that erythro-myeloid progenitor cells arise in the yolk sac at embryonic day 8.5. Two days later, they colonize the fetal liver. There, they give rise to red blood cells and progenitors of tissue-resident macrophages, including microglia, which migrate to their respective organs, including the brain (see Gomez Perdiguero et al., 2014). Just after the blood-brain barrier walls off the brain from new immigrants, hematopoietic stem cells colonize the liver and take over the job of producing red blood cells and monocytes until the bone marrow matures.
Asked how the budding macrophages know which organs to home to, Geissmann said the cells could be pre-programmed in some way, or they may randomly end up in certain tissues and then adapt to their new environment. Richard Ransohoff of Biogen Idec in Cambridge, Massachusetts, expressed the concept metaphorically: “You may decide to move to Florida because you already have a bikini, or perhaps you randomly find yourself in Florida and then go buy one.”
Whichever is correct, tissue-resident macrophages must take on roles suited to their environment. Chris Glass of the University of California, San Diego, reported that tissue-specific signals shape gene-expression patterns in the incoming macrophages by triggering the activation of thousands of enhancer sequences (see Gosselin et al., 2014). Glass initially compared gene-expression patterns in microglial cells to two different types of macrophage that live in the abdomen—large and small peritoneal macrophages. Using RNA sequencing, he found that the two subsets of peritoneal macrophage expressed similar patterns of genes; however, the microglia expressed about 800 genes at levels more than 16-fold higher than both types of peritoneal macrophage, and vice versa. For this reason, he suggested that the tissue environment plays a strong role in dictating these gene-expression profiles. In support of this idea, the genes likeliest to be specifically expressed in particular macrophage types shut down when Glass transferred the cells into tissue culture dishes.
Glass next asked how the cellular environment dictates these patterns of gene expression, hypothesizing that the activation of enhancer regions likely plays a part. In a previous study, Glass and colleagues had reported that lineage-specific transcription factors that switch on early in development bind to enhancer regions and prime them for full activation. Once the cell arrives in its environment, other transcription factors, turned on by signals in the local milieu, bind adjacent to those poised enhancers, and turn on downstream genes. This two-hit scenario explains how gene activation can be specific to both a cell type and a unique environment (see image above and Heinz et al., 2013).
Glass hypothesized that the local signals in the peritoneum could be retinoic acid and in the brain, TGF-β, and that they might activate transcription factors in macrophages that would bind to PU.1-primed enhancer regions in the DNA. PU.1 is a macrophage-specific transcription factor. To test this idea, Glass first surveyed the landscape of enhancer and promoter activity throughout the genome by measuring histone methylation and acetylation, which usually coincide with areas where genes are active. Eight thousand promoters were marked in macrophages. The activity of about 300 of those was higher or lower in microglia compared to peritoneal macrophages. Starker differences emerged at enhancers. Glass found that about 50,000 were primed in macrophages and about a quarter of those were either more or less active in specific cell types. Glass also found evidence for around 500 “super-enhancers,” which, he said, are regions containing clusters of enhancers that work together to activate a single gene. Of those, around half were differentially activated between microglia and peritoneal macrophages.
Using a variety of biochemical and genetic techniques, Glass determined that signal-dependent transcription factors, such as SMAD (which is triggered by TGF-β in the brain), activated a subset of PU.1-poised enhancers and super-enhancers in microglia. For peritoneal macrophages, retinoic acid receptors elevated the activation of other PU.1-poised enhancers.
If the local environment plays such a major role, then could it effectively turn infiltrating monocyte-derived macrophages into microglia once they enter the brain? Researchers have been struggling to establish the provenance of macrophages and microglia in the brain. Glass speculated that because monocytes derive from hematopoietic stem cells in the bone marrow, rather than the yolk sac as microglia do, they would have some unique lineage-dependent transcription factors that would always make them a bit different than microglia. How different, and which factors stably indicate which lineage, is still up for debate.
Researchers at the meeting were impressed with Glass’s work, but also pointed out that the epigenetic findings need to translate into functional outcomes before it’s clear how important they really are. “These findings represent differences only in the ‘phenotypic potential,’ of macrophages,” commented Anne La Flamme from Victoria University of Wellington in New Zealand. Glass acknowledged that this is a problem that plagues all genetic and epigenetic studies. He sees epigenetics as a way to sift out what may be most important to the identity of each cell type.—Jessica Shugart
Microglia in Disease: Innocent Bystanders, or Agents of Destruction?
Researchers have long sought to understand what makes the microglial cells residing in the brain different from macrophages that live in other tissues or cruise the blood. Even more elusive is how microglia respond to disease. At “Neuroinflammation in Diseases of the Central Nervous System,” a Keystone Symposium held January 25-30 in Taos, New Mexico, researchers took a renewed stab at classifying these cells in the healthy body. They also attempted to track how the cells change in the face of ongoing pathology.
Chris Glass of the University of California, San Diego, reported that transcription-factor binding to vast landscapes of enhancer sequences could dictate how microglia express different sets of genes than macrophages do (see Part 2). How do these enhancer landscapes affect microglial gene expression, and how does disease change this? Oleg Butovsky of Harvard University dove into these questions in a data-packed talk. Butovsky used microarray and proteomics approaches to compare the gene expression signature of microglia, specifically CD45lo, CD11b+ cells from adult mouse brains to those of other brain cells and monocytes isolated from the spleen. He found that 239 genes were uniquely expressed in microglia compared to other macrophages and immune cells; of those, 106 genes were uniquely expressed in microglia compared to other cells in the brain. Because this gene-expression pattern occurred under healthy conditions, Butovsky dubbed it a “homeostatic signature” of microglia (see Butovsky et al., 2014).
At the Heart of Microglia.
Microglia have a unique pattern of gene expression that distinguishes them from other cells in the brain (pictured above) as well as from macrophages in other parts of the body.
[Image courtesy of Butovsky et al., Nature Neuroscience 2014.]
Interestingly, TGF-β was the most important upstream regulator of this homeostatic signature, said Butovsky. Adding TGF-β preserved the signature in cell culture; on the flip side, microglia were largely absent from the brains of TGF-β-deficient mice.
Butovsky next tested what would happen to this homeostatic signature in disease. In mouse models of multiple sclerosis, amyotrophic lateral sclerosis, and Alzheimer’s, microglia lost the signature and took on a more pro-inflammatory phenotype. What caused the signature to disappear when things got tough? Butovsky said that in every disease model he tested, microglia upregulated expression of ApoE as well as miR-155, and that both of these were required to erase the signature. The researchers looked for a specific inflammatory signal that boosted ApoE and miR-155. They found nothing. Finally, they discovered that something about dead neurons did the trick. “This is the one thing all of these diseases have in common,” Butovsky said. While he does not know how dead neurons eliminate the signature, he believes ApoE upregulation occurs upstream of miR-155 because microglia from ApoE knockout mice preserves the homeostatic signature. Blocking either ApoE or miR-155 also retains it.
It is unclear how the loss of the homeostatic signature changes the function of microglia in all disease scenarios. Butovsky recently published his finding that blocking miR-155 in a mouse model of ALS restored the microglial signature and ameliorated disease (see Butovsky et al., 2015). He is collaborating with MiRagen, a biotech company in Boulder, Colorado, to develop ALS treatments by blocking miR-155.
Richard Ransohoff of Biogen Idec in Cambridge, Massachusetts commented that it is important to remember that the microglial signature is based entirely on comparison with other cell types. “That doesn’t degrade the value of having a signature, but it means that the interpretation of the signature should be that it’s an operational, somewhat artificial construct to capture a subset of genes that are relatively characteristic of microglia as compared to other cells. It is not necessarily an indicator of the most important genes a microglial cell expresses, as some of those may be shared with other cell types, such as neurons or other types of macrophages,” he said.
In his presentation, Ransohoff also parsed microglial and monocyte differences. He analyzed the cells in a mouse model of multiple sclerosis—a disease in which peripheral monocytes invade the CNS. Ransohoff reported that in this scenario, brain microglia turned down expression of thousands of genes, whereas infiltrating macrophages ramped up expression overall. Chemokines, which attract other cells, were among the few genes that microglia did turn on in response to disease, suggesting they may have been beckoning macrophages from outside the brain. “Essentially, they shut down and scream for help,” Ransohoff said.
He added that in the case of MS, infiltrating macrophages seem to be the “bad guys” that cause damage, whereas microglia appear to be paralyzed bystanders. He found that the former appear to strip myelin off axons, while microglia only participate in cleaning up the resulting mess (see Yamasaki et al., 2014). These roles may be different in Alzheimer's, where the influx of infiltrating monocytes is less robust, Ransohoff emphasized. However, based on recent data generated in his and other labs, he hypothesized that in AD, infiltrating macrophages enter the brain and surround plaques, while microglia may sit idly by, likely inhibited by Aβ (see Krabbe et al., 2013).
What makes the microglia so passive? Ransohoff teamed up with Glass to determine how CX3CR1 (aka fractalkine receptor) might tie in. Microglia are the only cells in the brain that express this receptor. CX3CR1 knockout mice have a mixture of phenotypes. When crossed to AD models, they have fewer amyloid plaques, but when crossed to tau-transgenic mice, they have more tau pathology (see Oct 2010 news). Ransohoff and Glass examined microglia from CX3CR1-deficient mice, and found that pro-inflammatory genes—such as NLRP3 and NFκb subunits—were more active than in microglia from normal mice. More extensive enhancer studies are yet to come, but Ransohoff said the preliminary data suggest that CX3CR1 may be important for keeping microglial inflammation in check.
At Keystone, microglia inhabiting the brain’s gray matter and living in close communication with neurons drew the lion's share of attention. However, Marco Prinz of the University of Freiburg in Germany identified a novel pathway in microglia that inhabit white matter. He reported that only those microglia express USP18, a ubiquitin protease that regulates inflammatory responses. When Prinz knocked out this gene, either in a whole mouse or only in microglia, the microglia produced pro-inflammatory cytokines and destroyed the myelin encasing nearby axons. Prinz said that USP18 prevents this destruction not through the actions of its catalytic domain, but by directly interacting with Type I interferon receptors. Somehow, the interaction between the two molecules keeps the cells from responding to low levels of interferon that are consistently present in the brain, Prinz said.
The findings indicate that while microglia are often lumped together as a group of cells that differ from macrophages in other parts of the body, they are not monolithic but have diverse functions even among themselves, depending on where in the brain they are. “Prinz’s work demonstrated that microglia respond to very local signals within brain microenvironments,” said Hugh Perry of the University of Southampton, U.K. The work is an example of the ever-increasing complexity and specificity that researchers must contend with as they study the cells that mediate neuroinflammation, Perry added.—Jessica Shugart
Butovsky O, Jedrychowski MP, Cialic R, Krasemann S, Murugaiyan G, Fanek Z, Greco DJ, Wu PM, Doykan CE, Kiner O, Lawson RJ, Frosch MP, Pochet N, Fatimy RE, Krichevsky AM, Gygi SP, Lassmann H, Berry J, Cudkowicz ME, Weiner HL.
Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice.
Ann Neurol. 2015 Jan;77(1):75-99. Epub 2014 Nov 27
PubMed.
Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, Wu PM, Doykan CE, Lin J, Cotleur AC, Kidd G, Zorlu MM, Sun N, Hu W, Liu L, Lee JC, Taylor SE, Uehlein L, Dixon D, Gu J, Floruta CM, Zhu M, Charo IF, Weiner HL, Ransohoff RM.
Differential roles of microglia and monocytes in the inflamed central nervous system.
J Exp Med. 2014 Jul 28;211(8):1533-49. Epub 2014 Jul 7
PubMed.
United in Confusion: TREM2 Puzzles Researchers in Taos
Ever since genetic studies identified TREM2 as a risk factor for Alzheimer’s and other neurodegenerative diseases in 2012, researchers have raced to understand why. The trouble thus far has been that findings conflict as often as not, and the results presented at “Neuroinflammation in Diseases the Central Nervous System,” a Keystone Symposium held January 25-30 in Taos, New Mexico, were no exception. At first glance, data from different labs appeared to disagree on which cells expressed TREM2 and how they affected amyloid plaques. However, as the meeting progressed, researchers found common ground and charted plans to clarify lingering discrepancies. They seemed to agree that TREM2 channels supportive signals to myeloid cells surrounding plaques in AD model mice, but what those cells are and what they do remained controversial.
Meeting co-organizer Chris Glass of the University of California, San Diego saw the TREM2 controversy as a teachable moment for the field. As researchers learn more about this receptor, they will likely reveal fundamental realities about which aspects of neuroinflammation are protective, and which are harmful. “TREM2 is a perfect example of how little we know about neuroinflammation, and sorting out its function will be extremely instructive,” he said.
As its name suggests, TREM2 (which stands for triggering receptor expressed on myeloid cells) primarily adorns myeloid cells, including microglia and macrophages elsewhere in the body. No one knows what triggers it, but ligands ranging from lipopolysaccharide to mitochondrial chaperones to apoptotic neurons have been proposed (see Yaghmoor et al., 2014). When activated, TREM2 seems to soothe inflammation and promote phagocytic activity in macrophages and microglia (see Hickman et al., 2014). Multiple variants of the gene have popped up in genetic studies of neurodegenerative disease, and the R47H variant triples a carrier’s risk of AD (see Nov 2012 news). To understand how TREM2 and neuroinflammation influence AD, researchers have generated TREM2-deficient mouse strains and crossed them to different AD mouse models. The results—some presented for the first time at the meeting—sparked lively debate at Keystone.
Myeloid infiltrators?
TREM2-positive cells (green) also express CD45 (pink), a marker of infiltrating macrophages. These cells cluster, most likely around plaques, in APP/PS1 mice, whereas cells carrying P2RY12 (red), a marker of brain-resident microglia, steer clear. [Image courtesy of Taylor Jay.]
Taylor Jay was the first to put forward TREM2 data at the meeting. A graduate student in the labs of Bruce Lamb and Gary Landreth at Case Western Reserve University in Cleveland, Jay and colleagues had presented their findings at the Society for Neuroscience annual meeting in D.C. last November (see Dec 2014 conference coverage). Jay reported that TREM2-expressing cells surrounded amyloid plaques in 4-month-old APP/PS1 mice. These cells highly expressed CD45, a marker of peripheral macrophages, and little P2RY12, a marker associated with microglia. These macrophages were absent from the brain in TREM2-deficient APP/PS1 mice. On the other hand, resident microglia (singled out by their low expression of CD45 and high expression of P2RY12) expressed no TREM2. They remained just as evenly spaced throughout the brain in APP/PS1 mice as they were in controls, indicating they were not the cells gathering around plaques. Jay dropped a counterintuitive bomb when she reported lower plaque burden in the hippocampus of TREM2-deficient mice.
Microglia Mosaic.
In TREM2-negative APP/PS1 mice, microglia (green), maintain their regular tiling pattern, even in the face of amyloid plaques (red). [Image courtesy of Taylor Jay.]
Researchers galore lined up behind the microphones to pepper Jay with questions after her talk. V. Hugh Perry of the University of Southampton in England seemed perplexed that resident microglia weren’t the ones expressing TREM2, as previous studies had suggested they did. Jay responded that in their hands, microglia indeed expressed TREM2 RNA transcripts, but not a detectable level of protein. Oleg Butovsky of Harvard University, who originally generated the antibody to P2RY12, commented that the marker was an unreliable way to label resident microglia in APP/PS1 mice because its expression plummets in the context of disease. Jay said the combination of other markers she used to identify the cells made her confident she was looking at microglia. Finally, Arnon Rosenthal of the biotech company Alector in San Francisco brought up the elephant in the room: How could the plaque burden Jay had presented explain data suggesting that loss of TREM2 function raises AD risk? The findings were indeed perplexing, Jay acknowledged. Perhaps generating TREM2 knock-in mice that harbor disease-associated mutations could help, she suggested.
Marco Colonna of Washington University, St. Louis, presented data painting TREM2 as a microglial survival factor in AD. Colonna crossed TREM2-deficient mice with 5xFAD mice. When the crosses reached 8 months of age, their hippocampal plaque burden outpaced those in 5xFAD animals. Colonna saw no difference in the cortex. These findings conflicted with Jay’s; however, both researchers were quick to point out that they used different mouse models and measured plaques at different time points. Jay speculated that TREM2 could promote plaques early on, and then help remove them later. While that idea will need to be tested, some researchers believe that there may be stark differences between inflammatory responses that occur early and late in the disease process (see Eikelenboom and Hoozemans comment on Matarin et al., 2015). A recent study using postmortem brain samples reported that TREM2 expression levels are highest in brain regions most vulnerable to AD pathology, such as the hippocampus, and that expression rises as the disease progresses (see Strobel et al., 2015).
Colonna and colleagues next took a close accounting of the cells surrounding plaques. He told the audience that Iba1-positive cells, which he interpreted as resident microglia, surrounded plaques in 5xFAD mice. In TREM2-deficient 5xFAD mice, the total number of microglia in the brain dropped, and Colonna observed few cells surrounding plaques. Those that did expressed apoptotic markers. Except for the proposed origin of the cells, these findings agreed with Jay’s, who had also reported fewer cells around plaques in TREM2-deficient mice.
How might TREM2 boost the number of cells surrounding plaques? Colonna reported that TREM2 cooperated with colony-stimulating factor 1 receptor (CSFR1) to promote microglial survival. He hypothesized that when microglia proliferate in response to an insult such as amyloid plaques, they deplete colony-stimulating factor and thus may require co-signaling by TREM2 to amplify the signal. In support of this idea, Colonna reported that microglia isolated from TREM2-deficient mice or TREM2-deficient 5xFAD mice died off in culture when colony-stimulating factor levels were low, whereas cells expressing TREM2 managed to survive. All in all, Colonna suggested that TREM2 supports the survival of microglia during disease.
This left an obvious question: What activates TREM2? Colonna tantalized the audience by suggesting that phospholipids do. He used a reporter cell line that expresses green fluorescent protein (GFP) under control of the NFAT transcription factor. NFAT translocates to the nucleus in response to calcium mobilization when TREM2 is activated. Some anionic and zwitterionic lipids, including phospholipids such as phosphatidylserine, turned on GFP. TREM2 antibodies blocked this. Perhaps most intriguingly, many lipids failed to switch on GFP in cells that expressed TREM2 with the R47H high-risk variant. Unlike other TREM2 mutations that associate with neurodegenerative disease, R47H does not impair the receptor’s transport to the cell surface or shedding of its extracellular domain, although it does alter the protein’s glycosylation status (see Alzforum webinar and Park et al., 2015). The loss of an arginine residue in the R47H mutation would theoretically prevent TREM2 from interacting with anionic lipids, thus reducing microglial survival during disease.
In response to audience questions, Colonna speculated that lipids such as phosphatidylserine, which are exposed on the surface of dying neurons, could trigger TREM2 when disease strikes (see Takahashi et al., 2005). Sphingolipids released during demyelination could also trigger the receptor, and a recent study reported that TREM2 is required for efficient clearance of myelin debris (see Cantoni et al., 2015). Furthermore, lipids are known to associate with Aβ and facilitate aggregation. While Colonna found no evidence that Aβ directly triggers TREM2, he speculated that lipids associated with Aβ or exposed or released by Aβ-induced damage could. The loss of TREM2 expression did not impair microglial phagocytosis in vitro, but Colonna noted that increased cell survival would have the effect of enhancing any microglial function.
David Holtzman of WashU, who collaborated with Colonna on the work, said it will be interesting to test whether another major AD risk factor, that is, apolipoprotein E, plays a role in lipid binding by TREM2. As its name suggests, ApoE binds lipids, and the E4 isoform promotes accumulation of Aβ. Whether TREM2 and ApoE4 increase the chances of getting AD by affecting similar lipid-related pathways will be the subject of future experiments, Holtzman said. Interestingly, a recent study reported that the ApoE4 isoform exacerbates the microglial inflammatory response and turns down expression of TREM2 (see Li et al., 2015).
Holtzman’s group had previously reported no change in amyloid plaque burden in an APP/PS1 mouse heterozygous for TREM2 (see Jun 2014 news).
Given that three different groups have reported varying phenotypes of TREM2 deficiency related to plaques, Erik Musiek, also at WashU, commented that perhaps plaques are the wrong place to look. “We may have to look beyond our usual phenotype when assessing the function of TREM2,” he said. “If there were striking effects on plaques, I think it would be obvious.” Musiek noted that the meeting helped identify other potential neuroinflammatory phenotypes, such as microglial gene expression signatures (see Part 2 and Part 3 of this series). These may be useful in figuring out how TREM2 raises disease risk. “We need to start defining these signatures in the AD brain and find out which ones are important,” he said.
Regardless of whether cells gathered around plaques are infiltrating macrophages or resident microglia, scientists still need to figure out what they do and how TREM2 plays a role. Landreth hypothesized that Aβ tempers microglial phagocytosis and presented a way to change that. Landreth previously reported that bexarotene, a retinoid X receptor agonist, boosted phagocytosis and cleared plaques in mouse models of AD and even improved cognition, though other groups reported difficulty reproducing the results (see May 2013 news).
Appetite for Destruction?
Iba1+ macrophages (green) co-express TREM2 (blue) and MerTK (red), a receptor that promotes phagocytosis, in response to treatment with bexarotene. [Image courtesy of Julie Savage.]
At Keystone, Landreth reported that bexarotene increased the expression of Axl and Mer, two surface receptors known to enhance phagocytosis, on cells surrounding plaques in mice. These Axl/Mer-expressing cells also expressed TREM2 and high levels of CD45, suggesting they were infiltrating macrophages. Bexarotene treatment reduced plaque burden in the mice, and Landreth found that in vitro, bexarotene stimulated phagocytosis. Landreth interpreted the findings as further evidence that the cells surrounding plaques in AD are not resident microglia, but infiltrating macrophages that express TREM2.
Richard Ransohoff of Biogen Idec in Cambridge, Massachusetts, who collaborated with Landreth, Lamb, and Jay, sought to find common ground among the variety of TREM2 data at the conference. While Ransohoff remains convinced by Jay’s staining data that the cells surrounding plaques are macrophages that infiltrate from the periphery and Colonna prefers the idea that they are resident microglia, a commonality is that those cells disappear in AD models. Regardless of their origin, researchers agreed that what matters most is what those cells are doing around the plaques.—Jessica Shugart
Microglia may like to dine on amyloid plaques, but does the immunosuppressive environment in the brain quash their appetite? Apparently so. According to two recent studies, lifting the suppression unleashes the microglia’s craving, allowing them to feast on plaques, but without sampling neurons. The studies—one led by Todd Golde at the University of Florida in Gainesville and the other by Terrence Town at the University of Southern California in Los Angeles—used complementary approaches to reveal that the anti-inflammatory cytokine IL-10 worsens Alzheimer’s disease pathology and cognitive decline in mouse models. Removing the cytokine slowed disease pathology and kept synapses intact. Kevin Doty presented findings from the Town lab at “Neuroinflammation in Diseases of the Central Nervous System,” a Keystone symposium held January 25-30 in Taos, New Mexico. That work appeared in the February 4 Neuron, while Golde's was published in the January 7 issue of the same journal.
“It turns out that you can have your cake and eat it too,” Town told Alzforum. “Revving up microglia removes plaques without damaging neurons.”
Both labs had already shared preliminary results from their studies at conferences covered by Alzforum (see Mar 2013 and Dec 2013 conference coverage). In their published form, the studies also revealed that IL-10 boosts astroglial expression of ApoE, which the researchers reported decreases microglial appetite for Aβ in vitro. Other data presented at Keystone put ApoE as a major factor in promoting a pro-inflammatory microglial phenotype (see Part 3 of this series).
Neuroinflammation plays an important role in clearing the brain of debris, infectious pathogens, and sickly cells. However, if uncontrolled or chronic, it can also harm the brain, and this can occur during neurodegenerative disease (see Wyss-Coray and Mucke, 2002; Perry, 2010).
The quieter role of the yin—the anti-inflammatory cytokines—had been less explored. Two major anti-inflammatory cytokines, TGF-β and IL-10, are known to shut down immune responses before they spin out of control. In 2008, Town generated AD mice lacking TGF-β activity in macrophages and microglia. He reported that mice lacking this wet-blanket cytokine had fewer plaques and less cognitive decline than mice with TGF-β. Importantly, this occurred without damage to neurons (see Jun 2008 news). The result encouraged Town to go after IL-10.
In Town’s study, first author Marie-Victoire Guillot-Sestier and colleagues crossed IL-10-deficient mice with APP/PS1 mice. They found that year-old APP/PS1 mice harbored far fewer plaques when they lacked the cytokine. In biochemical analysis, soluble and insoluble Aβ40 and Aβ42 plummeted by more than half. In brain slices, activated microglia crowded around any remaining plaques, which appeared more diffuse than in animals with the cytokine. These cells expressed low levels of CD45, suggesting that they were resident microglia rather than infiltrating macrophages from outside the brain (see Part 2, Part 3, and Part 4 for more on the nature of immune cells in the AD brain). Immunostaining also revealed that these cells contained Aβ within phagolysosomes. When put into a dish, microglia harvested from IL-10-deficient mice more readily gobbled up aggregated Aβ42 than normal microglia did, and they sported more markers of activation.
Feeding Frenzy?
Activated microglia (red) from APP/PS1 mice lacking IL-10 (bottom) more readily clustered around plaques (green) than microglia from control APP/PS1 mice (top). [Image courtesy of Guillot-Sestier et al., Neuron, 2015.]
Importantly, despite their penchant for destroying plaques, microglia in IL-10-deficient mice left neurons alone. In fact, these mice had wild-type levels of the synaptic marker synaptophysin, suggesting that synapses were preserved. This paid off in some cognitive benefits. The mice more readily recognized novel objects (a signature of episodic memory) than APP/PS1 mice, and they were less hyperactive. However, IL-10 deficiency did not rescue APP/PS1 mice from deficits in spatial working memory.
To take stock of the changes wrought in the IL-10 deficient mice, the researchers compared their gene expression with that of APP/PS1 mice. They identified 117 differentially expressed genes, the majority involved in innate immune function, chemoattraction, Aβ interaction, or phagocytosis. Surprisingly, the researchers found that IL-10-deficient APP/PS1 mice made less ApoE.
Could a dearth of ApoE explain the microglia's taste for Aβ? ApoE influences clearance of different forms of Aβ, but it is not clear how (see Apr 2013 news). The researchers tested whether ApoE would affect microglial uptake of Aβ. ApoE2 barely altered uptake of Aβ aggregates, whereas ApoE3 and ApoE4 reduced it by about 70 percent. Importantly, IL-10-deficient microglia efficiently consumed Aβ when mixed with either ApoE2 or ApoE3, but not ApoE4 (see Alzforum Webinar).
In Taos, Gary Landreth of Case Western Reserve University in Chicago noted that it was unclear how ApoE was inhibiting Aβ uptake in this tissue culture model, and he questioned whether the experiments were modeling bona fide phagocytosis.
Golde’s lab took the reciprocal approach. Rather than knocking out IL-10, first author Paramita Chakrabarty and colleagues overexpressed the cytokine in the mouse brain. The researchers used a method they had previously pioneered to overexpress other cytokines. They injected adeno-associated virus 2/1 (AAV2/1) expressing IL-10 directly into the brain of neonatal CRND8 mice.
For the most part, the results of Golde’s study mirrored Town’s. Overexpression of IL-10 elevated plaque burden, damaged synapses, and accelerated cognitive decline. Microglia huddled around plaques but cleared little Aβ. A comparison of whole-brain gene-expression profiles revealed 140 genes differentially expressed between CRND8 mice with or without IL-10 overexpression, including chemokines, complement, and immune signaling genes. ApoE was upregulated 1.7-fold in CRND8 mice overexpressing IL-10.
As had Town’s group, Chakrabarty and colleagues decided to investigate the link between IL-10 and ApoE. The researchers found that ApoE protein levels rose nearly fourfold in the insoluble fraction of brain extracts of mice overexpressing IL-10, and immunofluorescence showed that ApoE was expressed by astrocytes and intermixed with plaques. To measure ApoE’s effects on Aβ uptake by microglia, the researchers incubated the cells in media taken from IL-10-deficient astrocyte cultures, which are chock-full of ApoE. When they fed the cells aggregated, fluorescent Aβ, they saw a reduction in microglial uptake compared to cells grown in normal astrocyte media.
Town said it was exceedingly rare that two papers agreed to this extent. “It’s really uncanny,” he said. Both Town and Golde propose a mechanism whereby microglia secrete IL-10, which then prevents them from taking up Aβ. In parallel, IL-10 boosts ApoE expression, which could reduce Aβ uptake even more.
Could blocking IL-10 work as a therapy for AD? Milan Fiala of the University of California, Los Angeles, found the results intriguing, but commented that blocking IL-10 in people would likely have inconsistent effects, as background levels of inflammatory and anti-inflammatory cytokines vary markedly throughout the population.
Richard Ransohoff of Biogen Idec in Cambridge, Massachusetts, agreed with Fiala, noting that blocking IL-10 could trigger harmful inflammatory effects. He added that results based on the manipulation of a single cytokine, such as IL-10, could be too simplistic to support therapeutic conclusions.—Jessica Shugart
Systemic Inflammation: A Driver of Neurodegenerative Disease?
Under healthy conditions, microglia look placid. They sit evenly spaced throughout the brain, processes extended, quietly doing their job of scanning for debris. When disease kicks in, these calm cells can transmogrify and end up doing more harm than good. As discussed at “Neuroinflammation in Diseases of the Central Nervous System,” a Keystone meeting held January 25-30 in Taos, New Mexico, the rabble-rousing signals that fire up microglia are not confined to the brain but also come from “below the neck,” said Hugh Perry of the University of Southampton in England. Whether triggered by acute infections or chronic disease, systemic inflammation may amplify microglial responses and exacerbate neurodegeneration, according to researchers at the meeting. They proposed ways to slow disease progression by soothing systemic inflammation.
The human body accumulates inflammatory battle scars as we age, whether through repeated assaults by microbial infections or chronic inflammatory diseases such as diabetes or atherosclerosis. Considering how systemic inflammation might alter the course of neurodegeneration is thus crucial, Perry said. “Old brains are attached to old bodies, and bodies tend to accumulate a lot of pathology over the years,” he said. At the meeting, Perry reported that this pathology primes microglia, making them prone to overreactions that could exacerbate neurodegeneration.
More than a dozen years ago, Perry and colleagues used a mouse model of prion disease to establish the idea that microglia can be primed. They found that microglia in prion-infected mice exhibited a state of heightened activation, and that the cells responded with extra gusto—pumping out pro-inflammatory cytokines and accelerating neurodegeneration—when the researchers subsequently challenged the mice systemically with lipopolysaccharide (see Combrinick et al., 2002).
Priming the Inflammatory Pump. In response to misfolded proteins or debris from neurodegeneration, microglia (blue cells) ramp up production of some proteins (left panel). When systemic inflammation strikes, perivascular macrophages (purple) and endothelial cells kick the primed microglia into overdrive (right panel). [Image courtesy of Perry and Holmes, Nature Reviews Neurology, 2014.]
The group has since reported that inflammation from inside or outside the brain can prime microglia (see Perry and Holmes, 2014). In one study, systemic infection with salmonella stimulated microglia to secrete pro-inflammatory cytokines and express surface activation markers for several weeks. Endothelial cells lining the blood-brain barrier (BBB) also responded to the systemic alarm by expressing adhesion molecules. Subsequent injection of lipopolysaccharide into the brains of primed mice triggered much higher levels of microglial activation than in mice that hadn’t been infected (see Püntener et al., 2012).
Perry hypothesized that similar triggering scenarios must happen in the human brain throughout life, and that this would affect the progression of neurodegeneration. In collaboration with Clive Holmes at the University of Southampton, the researchers tracked cognitive symptoms in 300 people with AD over a six-month period. They found that cognition in patients who came down with a systemic infection declined twice as fast as it did in those who didn’t. People with chronic inflammation, as measured by high baseline levels of the proinflammatory cytokine tumor necrosis factor alpha (TNFα), declined at quadruple the rate of those without (see Holmes et al., 2009).
Based on this study, Holmes and Perry hypothesized that blocking TNFα would slow disease. At Keystone, Perry presented the results of a small pilot study that tested this. Over six months, the researchers treated 20 AD patients with weekly subcutaneous injections of etanercept, a decoy receptor that binds TNFα. The researchers monitored the patients’ performance on neuropsychiatric tests and activities of daily living. They found that while 20 other AD patients getting a placebo tended to decline in these measures over the study period, those taking the TNFa blocker stabilized. “As a pilot study, it shows you that the arrow points in the right direction,” Perry said. He stressed that larger studies will be needed to see if the effect holds. The results offered a glimmer of hope that targeting inflammation could have therapeutic potential, especially following disappointing results of non-steroidal anti-inflammatory drugs (NSAIDs) for AD (see May 2008 news).
Malu Tansey of Emory University in Atlanta asked Perry if he thought etanercept could affect AD progression by working systemically, or by eking into the brain in small amounts. Perry said there was scant evidence that the drug enters the brain at all, so it was likelier that any effects on AD would occur by quelling systemic inflammation.
Kathryn MacPherson, a graduate student in Tansey’s lab, presented initial findings from a study of a more selective TNFα inhibitor, XPro1595. TNFα exists in two forms that have opposing effects. A soluble form triggers pro-inflammatory responses that promote chronic inflammation and cell death, whereas the transmembrane version of the protein facilitates neuroprotective responses (see Montgomery and Bowers, 2011). Unlike etanercept, which blocks both forms of TNFα, XPro1595 only blocks soluble TNFα. The inhibitor also enters the brain.
MacPherson reported that 5xFAD mice display age-dependent brain inflammation. Treating the mice with XPro1595 reduced the number of activated, CD45-hi myeloid-derived cells (presumably infiltrating monocytes) in the brain, and also altered T-cell populations there. While the inhibitor’s effects on plaques have not yet been measured, MacPherson reported that in studies performed in collaboration with Chris Norris at the University of Kentucky, it rescued deficits in synaptic strength and long-term potentiation in 7-month-old 5xFAD mice. The researchers did not observe an elevation in systemic inflammation in 5xFAD mice as compared to non-transgenic mice, suggesting that XPro1595 may work by calming inflammation in the brain. To measure the drug’s effects in a situation more akin to aging people with chronic systemic inflammation, MacPherson plans to investigate the effects of the inhibitor in AD mice on a high-fat or high-sugar diet. FPRT, a small biotech company, is developing the drug for the treatment of neurodegenerative diseases, and it reduces cell loss in a rat model of Parkinson's (see Barnum et al., 2014).
Glenn Rall of Fox Chase Cancer Center in Philadelphia presented another example of cross-talk between immune responses in the brain and the rest of the body. Rall detailed how T cells responding to a virus in the periphery can move into the brain and wreak havoc there. He studied mice systemically infected with lymphocytic choriomeningitis virus (LCMV), which evokes a strong CD8+ T cell response but does not infect the brain. While this virus raged, he injected mice intracranially with a measles virus that only infects neurons in the CNS. The mice survived infection by either of the two viruses alone without even getting sick, but co-infection killed half of them. The mice died because LCMV-specific CD8+ T cells from the periphery infiltrated the brain, where they triggered deadly edema. These cells stayed out of the brain of mice infected only with LCMV, suggesting that the CNS measles infection set the stage for the disastrous recruitment (see Matullo et al., 2011). Rall found that other virus combinations, such as systemic ectromelia combined with CNS-restricted polio, triggered a similar infiltration of T cells.
Could a systemic infection set the stage for T-cell incursion into the brain even after the illness has passed? To test this, Rall infected mice systemically with LCMV, waited a month (long after the virus was cleared), and then infected their CNS with measles. These mice became ill, unlike controls that received only the measles virus. Rall detected T-cell infiltration in this model as well, but has not yet confirmed those T cells were specific to LCMV.
Although T cells were the agents of destruction in Rall’s co-infection model, he plans to investigate whether microglia or infiltrating macrophages were instigators. It’s possible that primed macrophages beckoned the T cells into the brain, Rall said.
Perry was fascinated by the idea that CNS infection opened the brain to infiltration by peripheral immune cells. He said that in his salmonella infection model, peripheral T cells also infiltrated the brain, even though the BBB remained intact. He speculated that activated endothelial cells within the BBB somehow facilitated this recruitment.
Along those lines, Richard Daneman of the University of California, San Diego, focused on structural changes that weakened the blood-brain barrier in disease. Although endothelial cells that make up the BBB are akin to those that line other vessels in the body, BBB endothelial cells form a much tighter barrier. In a previous study, Daneman and colleagues found that BBB endothelial cells maintain this barrier in response to signals from pericytes. The latter lie on the parenchymal side of the BBB and communicate with brain-resident cells and the endothelium. Pericytes prevent BBB endothelial cells from expressing “leaky genes” expressed by endothelial cells in other parts of the body (see Daneman et al., 2010). At Keystone, Daneman reported that one of these genes, EHD4, regulates endothelial permeability. Normally, BBB endothelial cells do not express this vesicular protein, but in mouse models of stroke, multiple sclerosis, traumatic brain injury, and epilepsy, or in pericyte-deficient mice, they upregulate EHD4. When Daneman overexpressed EHD4 specifically in BBB endothelial cells, he observed that the protein localized within vesicles of the cells, where it facilitated the transcytosis of proteins normally barred from the brain, such as fibrinogen.
Daneman proposed that during disease, pericytes lose their connections with endothelial cells, lifting suppression of EHD4 and other genes that promote unintended transport across the BBB. Daneman has so far tested this theory in CNS disease models, but researchers at the meeting wondered if the same breakdown happens during systemic inflammatory diseases, such as lupus or atherosclerosis. If this were the case, targeting EHD4 and related pathways could offer a way to prevent barrier disruption in many diseases, Daneman said.
Would such barrier disruption promote the damaging cross-talk that Perry and Rall saw in their models of microglial priming and T-cell infiltration? Perry told Alzforum that so far, evidence suggests that the communication between the periphery and the brain occurs across an intact BBB in these models, although he offered that a transient breakdown in the BBB could facilitate enhanced infiltration of inflammatory mediators. Rall agreed. “A leaky barrier is indiscriminate in terms of what it allows into the brain,” he said. “I think these immune cells are crossing capillaries with an intact barrier, due to an activated endothelium.”
The integrity of the BBB, and how it relates to communication with the rest of the body during disease, remains an area of active debate. For example, at Keystone, Ryan Watts of Genentech in San Francisco suggested that the barrier remains intact in mouse models of AD, as well as in tau transgenic and ApoE-deficient mice. Watts measured transport of antibodies across the BBB in these mice and found it was no different from wild-type. Human disease may be a different story, however. Researchers recently reported subtle leaks in the hippocampus of older people and in people with mild cognitive impairment, though this finding has not yet been independently replicated (see Alzforum Webinar).
Whether the flow of immune communication is haphazard or carefully orchestrated, researchers seemed to agree that the brain and the body share the burden of inflammation during aging.—Jessica Shugart
Microglia Rely on Mixed Messages to Select Synapses for Destruction
Microglia may be the brain’s trash collectors, but when it comes to neurons, they are quite discerning, it turns out. At “Inflammation in Diseases of the Central Nervous System,” a Keystone symposium held January 25-30 in Taos, New Mexico, researchers reported that the cells interpret a combination of go/no-go signals to select synapses for disposal. Researchers raised the possibility that the signals go haywire in neurodegenerative diseases such as Alzheimer’s, and proposed that revving up protective signals or shutting down destructive ones could point to therapeutics.
Beth Stevens of Children’s Hospital, Boston, reported that synaptic strength may govern which synapses are picked off by microglia, and which survive. As a postdoc in Ben Barres’s lab at Stanford University in California in 2007, Stevens reported that neuronal circuitry was streamlined during development by the complement cascade—a multi-protein defense system that tags pathogens and debris for destruction. The complement protein C1q, expressed in the CNS during development, tagged the surface of synapses and triggered their pruning as well (see Stevens et al., 2007). C1q initiates the complement cascade, a proteolytic sequence of events that results in the cleavage of the complement protein C3. Products of C3 cleavage then bind to the surface of the tagged synapse and beckon microglia, which express the C3 receptor, to eat them. Since helming her own lab, Stevens has begun to uncover how this cascade results in the selection of certain synapses for destruction, while sparing others.
In 2012, Stevens reported that microglia preferentially target synapses that transmit the weakest signals to postsynaptic neurons. By injecting different fluorescent tracers into the eyes of mice, Stevens traced axonal projections from neurons in the retina to those in the visual cortex. When Stevens inhibited neuronal activity in one eye and not the other, she saw that microglia preferentially engulfed synapses from neurons in the blocked eye. It appeared that microglia were somehow singling out the weakest synapses for destruction (see Schafer et al., 2012). Last month in Taos, Stevens reported that this predilection for weaker synapses is dependent on C1q, suggesting that the complement cascade directs microglia to devour synapses.
To scrutinize the mechanisms behind pruning selection at the single synapse level, researchers in the Stevens lab persevered for five years to construct an in vitro neuronal circuit system. They based it on connections that form between retinal neurons that make up the optic nerve, and neurons in the dorsolateral geniculate nucleus (dLGN), a major relay station in the thalamus that receives inputs from the optic nerve and passes them onto other regions of the brain. They co-cultured two pieces of retina (each labeled with a different dye), along with a piece of embryonic dLGN from a mouse embryo. In vitro, the retinal ganglion cells sprouted axons that projected into the dLGN, where they formed synapses that underwent pruning after some time in culture.
Leaving this circuit unperturbed, axons from both slices of retina innervated most of the dLGN neurons. However, when the researchers increased synaptic transmission in one retina, synapses onto dLGN neurons from this more-active retina quickly outnumbered those from the less-active retina. Stevens said microglia in the cultures likely eliminated the weaker synapses, but only future research will confirm that. Interestingly, complement dictated this competition, because when the cultures were prepared from C1q-deficient mice, activated or inhibited retinal ganglion cells equally innervated the dLGN. Stevens said her lab will continue to use this new tool to focus in on selection at the level of individual synapses.
In addition to the destructive signal delivered by C1q, Stevens said her lab was investigating a potential protective signal that discourages microglial pruning. She plans to use the lab’s retinal culture model to determine how the two signals interact, and whether the protective signal is expressed on some synapses and not others.
Carla Shatz of Stanford University presented recent findings about PirB, another regulator of synaptic pruning. As previously covered by Alzform, Shatz reported that the major histocompatibility complex class I (MHCI) receptor is expressed on neurons, and keeps synaptic plasticity in check by promoting synaptic pruning. Mice that lack the receptor display enhanced ocular dominance, a classic example of neuronal plasticity in which neurons emanating from an open eye form more synaptic connections than those projecting from a closed one. Interestingly, Aβ activates PirB, kicking pruning into overdrive (see Sep 2013 news). Shatz proposed that blocking the receptor could be a way to prevent the synaptic destruction that occurs in the early stages of AD, even before plaques appear.
To this end, Shatz and colleagues generated a soluble version of PirB to act as a decoy to distract Aβ and other ligands and block activation of PirB signaling on neurons. Injecting the decoy directly into mouse brains not only drove ocular dominance plasticity during development (when the process normally occurs), but in adult mice as well. When the researchers covered a single eye in mice for their first six weeks of life, normal mice were left permanently blinded in that eye, whereas PirB-deficient mice gradually regained much of their vision (see Bochner et al., 2014). Shatz plans to test whether blocking PirB in early stages of AD could fend off cognitive decline associated with synaptic destruction. Prompted by a question from the audience, Shatz said that the PirB-deficient mice outperform normal mice on nearly all cognitive tasks tested so far.
Marco Colonna of Washington University School of Medicine in St. Louis was intrigued by the expression of PirB on neurons, and asked if it was the only member of the large Pir protein family to be expressed on those cells. Colonna was among the first researchers to discover the human homologue of PirB, Lilrb2 (also known as ILT4), and had reported the receptor’s expression on peripheral myeloid cells (see Colonna et al., 1998). Shatz said that PirB is the only Pir protein detected on neurons, and added that according to the Allen Brain Atlas, Lilrb2 is expressed in the human brain.
From Development to Disease
Could overzealous synaptic disposal contribute to AD? Soyon Hong of the Stevens lab addressed that question in her Keystone talk. Because evidence points to synaptic loss due to Aβ oligomers as an early step in AD pathogenesis, Hong set out to measure whether the complement-mediated pruning that Stevens documented during development revs up again at the earliest stages of disease (see Selkoe, 2002; and Koffie et al., 2009). She looked for signs of complement-mediated synaptic pruning in the J20 model of AD. Hong found that a breakdown in synaptic connectivity in vulnerable regions of the brain preceded the appearance of plaques. This synaptic disconnection depended upon the complement cascade. The same process occurred in normal mice injected with Aβ oligomers, and correlated with microglial activation and their engulfment of some synapses. This dovetails with previous findings from both Stevens’ lab and Cynthia Lemere’s lab at Brigham and Women’s Hospital, Boston. It indicated that C3 deficiency preserved synapses and cognition in AD mice, even though these mice had a heavier plaque burden than their C3-expressing brethren (see Aug 2013 news).
Hong proposed that C1q latches onto neurons and triggers the complement cascade. This would result in the tagging of those neuronal synapses with C3, which triggers microglia expressing the C3 receptor to engulf synapses. Hong said she is investigating the relationship between Aβ oligomers and C1q binding to neurons.
Researchers at the meeting were impressed by the findings about synaptic pruning. “Both Shatz and Stevens’ data are exciting,” said Hugh Perry of the University of Southampton in England. “A few years ago, I was skeptical about the idea of this selective synaptic pruning, and wondered how it would work. Now, it seems there is a cohesive story coming together.” Perry added that more work remains to be done to ascertain whether selective pruning plays a role in human disease.
Shatz and Stevens plan to investigate how the mixed messages they identified—i.e., PirB, complement, and protective signals—blend together to target specific synapses for destruction, both in development and disease. Shatz told the audience that she hopes the research will one day lead to treatments that promote synaptic plasticity in aging people at a level closer to that achieved in the young. She added, jokingly, “I’m getting older and I want the pill.”—Jessica Shugart









Comments
No Available Comments
Make a Comment
To make a comment you must login or register.