Blessing or Curse? Peripheral Cytokines in the Brain
Quick Links
More friendly border than iron curtain, the blood-brain barrier lets all sorts of legitimate travelers pass. Could it also be letting some unsavory characters slip by? The subject of crosstalk between the peripheral circulation and the central nervous system permeated conversation at the 3rd Venusberg Meeting on Neuroinflammation, held 28 February-2 March 2013 at the Biomedical Center, University of Bonn, Germany. Researchers agreed that, in certain settings, circulating immune cells can enter the brain and create havoc (see ARF related news story). But they also emphasized that crosstalk occurs in the absence of infiltrating cells. Scientists argued that elevated plasma cytokines and other signaling molecules may profoundly affect the central nervous system, though not always negatively. While some proinflammatory cytokines might block neurogenesis in the brain, limit synaptic plasticity, or even cause cell death, others seem to encourage resident brain cells to take on protective roles, such as clearing amyloid-β plaques.
Scientists have studied links between plasma cytokines and the brain for decades. Charles Dinarello, University of Colorado, Denver, noted data from the Framingham Study that suggests people with the highest levels of circulating interleukin 1β (IL-1β) are almost three times more likely to develop Alzheimer's disease (see Tan et al., 2007). In contrast, people with more interleukin 1 receptor antagonist in their blood are less likely to have dementia, said Dinarello. He argued that peripheral cytokines, including IL-1β and other highly inflammatory ones such as IL-18, may predispose people to dementia and other degenerative conditions. IL-18, activated by IL-1β, drives apoptosis, and Dinarello showed that in a heart failure model, the IL-18 antagonist IL-18 binding protein (IL-18BP) rescues damage caused by IL-1β. Blocking IL-1β and IL-18 activation also limits damage in human myocardial tissue following ischemia (see Pomerantz et al., 2001). While these experiments implicate these cytokines in the heart, researchers have yet to dig into IL-18’s actions in the brain, said Dinarello.
Epidemiological evidence suggests a link. Dinarello noted an Italian study that found higher circulating IL-18BP in centenarians than in the general population, hinting that less free IL-18 promotes health (see Gangemi et al., 2003). People with AD have lower blood IL-18BP levels than age-matched controls, hence, higher free IL-18, as do people with certain systemic disorders. What these correlations mean was debated at the Venusberg meeting. Researchers noted it's difficult to separate cause and effect here, but Dinarello countered that emerging evidence points to peripheral cytokines profoundly influencing the central nervous system. "If they can get into the brain, they can cause damage," he said.
Many researchers at the meeting were intrigued by the link between systemic illnesses and dementia. Clive Holmes, University of Southampton, U.K., discussed an idea he has pursued for some time, namely that systemic inflammation can lead to central nervous system disease, including "sickness behavior," which resembles some of the neuropsychiatric symptoms of delirium, though it is a separate entity. Sickness behavior correlates with increased risk for AD (see ARF related news story). Holmes has reported that AD patients decline faster if they had a recent systemic infection, traumatic event (see Holmes et al., 2003), or elevated proinflammatory cytokines in their blood (see Holmes et al., 2009).
Animal models may help test correlations between circulating molecules and disease. Tony Wyss-Coray, Stanford University, Palo Alto, California, gave an update on his parabiosis model for studying blood factors that contribute to aging. In this model, two congenital mice share their blood systems, allowing Wyss-Coray to measure the effect of young blood on older animals, and old blood on younger. This led him to identify blood factors that appear to mediate aging, including the chemokine CCL11. Simply delivering CCL11 to young animals yields phenotypes associated with older mice, including less neurogenesis in the brain (see ARF related news story).
How else might blood factors influence tissue inside the CNS? In Bonn, Wyss-Coray said his group addressed this question by looking for differences in gene expression in hippocampal tissue from old/old and old/young parabiosis pairs. Researchers in his lab identified several gene networks that are altered in old animals exposed to young blood. The most highly upregulated turned out to be involved in synaptic plasticity. The group confirmed this at the protein level, looking at factors such as pCREB, Egr1, and cFOS. "The data indicate that factors in young blood reactivate the brain," said Wyss-Coray. In keeping with this, he reported greater synaptic spine density in older mice exposed to young blood.
How to achieve this effect without parabiosis? Wyss-Coray claimed that injections of young-mouse plasma improved cognition in older animals. "All this points to the possibility of some degree of rejuvenation, with neurogenesis, spine density, synaptic plasticity, memory, and inflammation all improving," he said.
But are any of these age-associated differences related to neurodegenerative disease? To test this, Wyss-Coray and colleagues compared gene expression and blood phenotypes among healthy controls and people with a semantic variant of frontotemporal dementia. Using pathway analysis software from Ingenuity Systems, they found elevated expression of networks involving interleukin 12 (IL-12) and IL-23 in the dementia patients. Independent research suggests that IL-12/23 signaling may drive autoimmunity, and exacerbate pathology and cognitive decline in mouse models of AD (see ARF related news story).
Wyss-Coray's group used a similar network-based approach to analyze blood phenotypes, measuring a panel of 50 proteins expressed by immune cells. The scientists found three subsets of markers more highly represented in plasma from FTD patients than controls. Some, including CD27, are known to contribute to hyperactivation of T cells. "Again, we seemed to have convergence around factors that contribute to autoimmunity," said Wyss-Coray.
What does this mean for FTD and other neurodegenerative diseases? For example, could ustekinumab, an IL-12/23 antibody currently used to treat the autoimmune skin disorder psoriasis, benefit dementia? Researchers at the Venusberg meeting were reluctant to speculate, but Wyss-Coray noted that autoimmune diseases were more common in FTD patients than would be expected by chance. Many of the affected patients have autoimmune skin diseases, but systemic lupus erythematosus and other disorders were represented as well.
For all the talk about cytokines and inflammation being bad for the brain, Terrence Town, University of Southern California, elaborated on a different idea, namely that of "good" neuroinflammation. Unlike mouse models in AD, Town’s transgenic rat model of AD recapitulates most cardinal pathologies of the disease, including Aβ accumulation, Aβ oligomers, gliosis, tau accumulation, synaptic loss, neuronal loss, and cognitive decline (see ARF related news story). In this model, microglial activation preceded obvious plaque formation, which hints that these cells may be attempting to prevent Aβ accumulation, said Town.
Town previously reported that blocking anti-inflammatory signals in peripheral macrophages promoted clearance of Aβ plaques in transgenic mice rather than exacerbating pathology, as might be expected if tipping the balance toward inflammation was bad (see ARF related news story). That result came from blocking TGF-β signaling. In Bonn, Town reinforced this finding by reporting that blocking other anti-inflammatory pathways had a similar effect. Town crossed PSAPP AD model mice with IL-10 and IRAKM knockouts. Both IL-10 and IRAKM quell inflammation, the latter being the only member of the interleukin 1 receptor-associated kinase family that suppresses innate immunity.
Both crosses accumulated fewer Aβ plaques than the PSAPP strain. They seemed to activate microglia, showing elevation of microglial markers Iba1+ and CD45 in the brain. In the IRAKM-negative animals, plaque burden fell while free Aβ rose in the brain, prompting Town to suggest that some dynamic process was going on to reduce plaques. He showed 3-D confocal microscopy reconstructions of brain tissue that demonstrated co-localization of Aβ with the phagolysosomal vesicle marker, LAMP1, demonstrating plaque phagocytosis by activated innate immune cells in vivo (see image below).
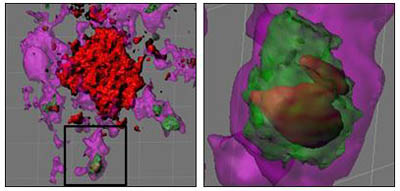
Glia Gobble Aβ
3-D confocal reconstructions of glial cells (purple) phagocytosing and digesting Aβ (red) via lysosomes (green). Images courtesy of Terrence Town and David Gate
“If it’s targeted, inflammation can be beneficial," said Town. "So we shouldn’t find it surprising that promoting specific types of microglial activation can lead to plaque clearance."—Tom Fagan.
References
News Citations
- Microglia Activation—Venusberg Meeting Questions M1, M2 Designations
- Alzheimer’s Risk Factors Roundup: Does Poor Health Hurt the Brain?
- Paper Alert: Do Blood-Borne Factors Control Brain Aging?
- Soothing Neuroinflammation Quells Plaques in Mice
- Honolulu: Monkey, Rat Join Menagerie of Mammalian AD Models
- Macrophages Storm Blood-brain Barrier, Clear Plaques—or Do They?
Paper Citations
- Tan ZS, Beiser AS, Vasan RS, Roubenoff R, Dinarello CA, Harris TB, Benjamin EJ, Au R, Kiel DP, Wolf PA, Seshadri S. Inflammatory markers and the risk of Alzheimer disease: the Framingham Study. Neurology. 2007 May 29;68(22):1902-8. PubMed.
- Pomerantz BJ, Reznikov LL, Harken AH, Dinarello CA. Inhibition of caspase 1 reduces human myocardial ischemic dysfunction via inhibition of IL-18 and IL-1beta. Proc Natl Acad Sci U S A. 2001 Feb 27;98(5):2871-6. PubMed.
- Gangemi S, Basile G, Merendino RA, Minciullo PL, Novick D, Rubinstein M, Dinarello CA, Lo Balbo C, Franceschi C, Basili S, D' Urbano E, Daví G, Nicita-Mauro V, Romano M. Increased circulating Interleukin-18 levels in centenarians with no signs of vascular disease: another paradox of longevity?. Exp Gerontol. 2003 Jun;38(6):669-72. PubMed.
- Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2003 Jun;74(6):788-9. PubMed.
- Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009 Sep 8;73(10):768-74. PubMed.
External Citations
Further Reading
News
- Microglia Activation—Venusberg Meeting Questions M1, M2 Designations
- Alzheimer’s Risk Factors Roundup: Does Poor Health Hurt the Brain?
- Paper Alert: Do Blood-Borne Factors Control Brain Aging?
- Soothing Neuroinflammation Quells Plaques in Mice
- Honolulu: Monkey, Rat Join Menagerie of Mammalian AD Models
- Macrophages Storm Blood-brain Barrier, Clear Plaques—or Do They?
- Inflammatory Crosstalk Between Periphery and Brain
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.