Joint Keystone Symposia: Neurodegenerative Diseases: New Insights and Therapeutic Opportunities and Neural Environment in Disease: Glial Responses and Neuroinflammation
At a joint Keystone symposia, microglia continued to twist and turn in the limelight, casting a long shadow as central agents of disease, not mere responders. Researchers embraced the complex reality of mixed pathology, dishing out data from animal models in which Aβ, tau, and α-synuclein mingled and sparked each other’s spread. From a gut-initiated model of Parkinson’s disease to chimeric mice harboring human microglia to a startling conversion of astrocytes to newborn dopaminergic neurons within the brain, researchers upped the ante on disease models and proposed new therapeutic strategies.
TREM2, Microglia Dampen Dangerous Liaisons Between Aβ and Tau
That microglia play a central role in neurodegenerative disease has become a premise; it’s no longer a question. The questions are all about the how. This was abundantly clear at joint symposia held June 16–21 in Keystone, Colorado. At Neurodegenerative Diseases: New Insights and Therapeutic Opportunities and Neural Environment in Disease: Glial Responses and Neuroinflammation, scientists exchanged their latest insights on how these exquisitely reactive glial cells shape the course of pathology, inflammation, and neurodegeneration. Via their surface receptor TREM2, microglia reportedly sculpt the architecture of Aβ plaques to lessen the formation of dystrophic neurites. Left to develop unchecked, these deformed neuronal extensions serve as fertile breeding grounds for tau aggregates, scientists found. Other researchers presented initial data from a chimeric AD mouse model flush with human microglia.
The preponderance of microglial genes among AD risk variants place these cells front and center in the disease. TREM2, the strongest among them, has been implicated in all manner of microglial functions. At Keystone, David Holtzman of Washington University in St. Louis placed TREM2 at the intersection between Aβ and tau pathology. As presented last year at Keystone by collaborator Christian Haass of the German Center for Neurodegenerative Diseases in Munich, and subsequently published, the scientists reported that without TREM2, mice were unable to rally microglia around Aβ plaques. This resulted in fluffier, more diffuse plaques infused with about half as much ApoE as those in mice replete with TREM2 (Jul 2018 conference news; Jan 2019 news). Holtzman also previously reported that in the PS19 model, which express the P301S mutant form of tau that causes frontotemporal dementia, TREM2 deficiency quelled harmful neuroinflammatory responses (Oct 2017 news).
This year, Holtzman put the two together, reporting how the microglia-plaque relationship affected the development of tau pathology. A graduate student in the lab, Cheryl Leyns, who is now at Merck, injected tau-laden material extracted from Alzheimer’s brains into the brains of 3-month-old APP/PS1 mice. A month later, she observed tau aggregates mingling with dystrophic neurites surrounding Aβ plaques, at least on the same side of the brain where she had made the injection.
Sluggish Recruitment. Microglia (green) rallied enthusiastically around plaques (blue) in mice expressing the common variant of TREM2 (left), but not the R47H variant (right). [Courtesy of Leyns et al., Nature Neuroscience, 2019.]
However, in APP/PS1 mice lacking Trem2, these dystrophic neurite-associated tau aggregates doubled. They even cropped up on the other side of the brain. A similar doubling of tau aggregates also occurred in mice expressing the R47H mutant form of human TREM2. These tau aggregates contained only endogenous mouse tau, suggesting they had formed by way of templated misfolding triggered by the injected human AD-tau.
Notably, the amount of aggregated tau correlated with the burden of dystrophic neurites in the brain, which, in turn, was higher in mice lacking functional TREM2. No tau aggregates had appeared when the researchers injected AD-tau before dystrophic neurites were forming, casting the damaged processes as a fertile substrate for subsequent seeding of tau. The findings appeared in Nature Neuroscience on June 24 (Leyns et al., 2019).
Sowing the Seeds of Tau. In a TREM2 knockout APP/PS1 mouse injected with AD-tau, tau aggregates (red) cluster around Ab42-laden (pink) plaques (blue). Microglia (green) hardly budge. [Courtesy of Leyns et al., Nature Neuroscience, 2019.]
Holtzman speculated that microglia tweak the architecture of plaques in a manner that requires TREM2 to function. His findings mesh with previous work led by Jaime Grutzendler of Yale University in New Haven, Connecticut. Grutzendler reported that without TREM2, microglia have trouble erecting a barrier around plaques, leading to “hotspots” of Aβ42 that wreak havoc on nearby axons (May 2016 news). Holtzman speculated that toxic tau could sprout from the ruins. “Dystrophic neurites, which are full of abnormal membranous material, may be a good substrate for allowing tau to convert to this irregular conformation,” he told Alzforum.
Haass believes that while this did not prove a connection between the two hallmark proteinopathies of AD, the results suggest as much. “Maybe the TREM2-dependent microglial function is the missing link that connects amyloid and tau pathologies,” he said.
Also at Keystone, David Hansen of Genentech in South San Francisco presented data that dovetailed with Holtzman’s. Hansen reported that, compared with PS2APP mice expressing TREM2, PS2APP mice lacking TREM2 had fluffier plaques and a higher proportion of Aβ42 to Aβ40. Hansen also reported an increase in LAMP1-positive, i.e. lysosome-packed, dystrophic neurites per plaque in TREM2 knockouts, and even found these damaged processes far away from plaques. Hansen’s group further examined structural tissue damage at the level of dendritic spines, reporting that TREM2 deficiency worsened spine loss near plaques.
What does all this mean for tau pathology? Hansen studied this in TauPS2APP model mice, which are PS2APP mice that express the P301L mutant form of tau. By nine months of age, TauPS2APP mice crossed to a TREM2-deficient background had more tau pathology than mice that did express TREM2.
Overall, Hansen drew similar conclusions to Holtzman: “Microglia, through TREM2, mitigate the degree to which Aβ pathology accelerates tau pathology,” he told the audience. Leyns, too, noted that together, both groups’ findings suggest that without TREM2, plaques assume a more Aβ42-laden form that damages neurons and creates a permissive environment for tau seeding.
Kim Green of the University of California, Irvine, reported that microglia not only compact plaques, but are required to build them in the first place. At Keystone, Green described what happened when he treated 6-week-old 5xFAD mice, which have no plaques yet, with the CSF-1R inhibitor PLX-5622. The treatment, which Green developed, depletes the mice’s microglia down to 2 percent of their normal numbers within a few days. Surprisingly to Green, as long as the mice’s chow contained PLX-5622 to keep microglia at bay, the mice developed barely any plaques. At 4 months of age, the few plaques that had grown were surrounded by those remaining few microglia.
Green fed PLX-5622 to the mice for as long as six months, eventually wiping out all their microglia. After that course, some plaques remained in the same regions where microglia had initially persisted with the shorter treatments, hinting the cells had been instrumental in building those plaques. Without microglia to compact them any longer, these remaining plaques had morphed into diffuse, fluffy deposits surrounded by dystrophic neurites, similar to those Holtzman observed. Green also reported that when PLX-5622 was initiated after plaques had formed, it did not affect overall plaque numbers.
Holtzman noted that while microglial depletion prevented plaques from forming in Green’s experiments, TREM2 deficiency merely altered the plaques’ architecture without strongly changing their overall burden in both his and Hansen’s work. This implies microglial factors other than TREM2 help build plaques, he said.
Green further reported that after six months on the inhibitor, the 5xFAD mice had large amounts of Aβ aggregate on the walls of their cerebral blood vessels, akin to cerebral amyloid angiopathy (CAA). Green suspects that without microglia corralling Aβ in the parenchyma, Aβ gets shunted into the vasculature instead. When the scientists stopped the inhibitor treatment and waited a month, microglia returned and plaques formed, though the CAA remained.
Green found no cognitive side effects of prolonged treatment with this drug, though some researchers wondered whether the CAA could cause cognitive deficits.
Green emphasized that he does not advocate wiping out microglia as a treatment in humans. After all, the cells have beneficial functions such as compacting plaques and limiting their neurotoxicity. However, he noted that in some scenarios, temporarily ridding the brain of stubbornly reactive microglia could theoretically allow new, less-damaging cells to repopulate it.
For example, a recent study reported that removal of reactive microglia, using PLX-5622, prevented “chemofog” triggered by methotrexate in mice. In that scenario, microglia riled up by the chemo drug derailed the development of oligodendrocytes, which led to deficits in myelination (May 2019 news). In another recent report, a necroptotic die-off of reactive microglia boosted remyelination in a model of multiple sclerosis (Jun 2019 news).
How fast do microglia rebound after PLX-5622 is gone, and where do the new cells come from? Monique Mendes, a graduate student in Anna Majewska’s lab at the University of Rochester in New York, addressed this question at Keystone. Using cranial windows to peer into the mouse brain before and after microglial depletion, Mendes saw that the few microglia that survived this treatment were the main suppliers of newborn microglia once the inhibitor was no longer there. Between two and four days after the mice stopped taking the drug, Mendes spotted microglia with two somas, some of which ultimately split into two cells. Only some microglia appeared destined for rapid division. While some microglia divided at a blistering pace—turning over within four to 12 hours per division—others remained trapped in a binucleate stage, apparently hesitant to split into two independent cells.
This data do not address whether microglia born into neurodegenerative environs might take on the reactive stance of their forebears, but Green told Alzforum that the finding jibes with a rapid repopulation his lab has observed as well. According to Mendes’ data, depleting 100 percent of microglia would preclude their recovery after treatment stops. Green told Alzforum that he will soon share data about what happens when every last microglia is depleted from the brain.—Jessica Shugart
Down to Sex? Boy and Girl Microglia Respond Differently
Some microglia are from Mars; others from Venus. Well, no. But in some scenarios, to which sex a microglial cell belongs appears to hold some sway over how it responds. Consider tau. At a joint Keystone symposia—Neurodegenerative Diseases: New Insights and Therapeutic Opportunities, and Neural Environment in Disease: Glial Responses and Neuroinflammation—Li Gan reported that female tauopathy mice expressing an AD risk variant of TREM2 exhibited a stepped-up microglial reaction and had worse memory. The same TREM2 mutation did no such thing in male mice.
Gan, who is now at Weill Cornell Medical College in New York, also uncovered profound differences in how microRNAs control gene expression in males versus females. Other researchers found that microglia dictate pain sensitivity in sex-specific ways. In all, the findings cast microglia as pulling the strings behind some sex differences. At least in mice.
Many presentations at Keystone focused on these resident immune cells and TREM2. To name a few, David Holtzman of Washington University, St. Louis, David Hansen, Genentech, and Kim Green, University of California, Irvine, implicated TREM2 and other microglial factors in shaping Aβ plaques, which influenced the subsequent development of tau pathology (see Part 1 of this series). But does TREM2 also affect tau independently of Aβ? On this question, Gan reported surprising results in the P301L model of tauopathy. Her lab made use of a menagerie of mouse lines, including TREM2 knock-ins in which one or both copies of the mouse gene were replaced by the human one. The knock-ins expressed either the common variant of human TREM2, or the R47H-TREM2 variant that boosts risk for AD, and the animals were generated on the wild-type or P301L background. In addition, Gan examined P301L mice on a TREM2 knockout or wild-type background. This is where sex differences became apparent.
Compared to 1-year-old wild-type mice expressing a copy of normal human TREM2, those carrying a copy of the R47H AD variant had a spatial-learning deficit. It was greater in female than male mice. This sex difference grew further when Gan crossed the TREM2 mice to a P301L background. In Keystone, she reported that P301L mice expressing one copy of R47H-TREM2 had the same burden of tau pathology as mice expressing normal mouse or human TREM2. That said, female P301L mice expressing the AD risk variant did more poorly at spatial learning, whereas the mutation appeared to slightly benefit performance of male P301L mice.
Gan pegged maladaptive microglia as a likely culprit behind the greater memory loss in female P301Ls. She reported that microglia in both male and female P301L mice expressed the disease-associated microglia (DAM) signature of genes. However, expression of certain DAM genes shifted only in female mice that expressed TREM2-R47H. In females, R47H dialed up expression of many pro-inflammatory cytokines of the DAM signature and lessened expression of a suite of neuronal genes. In male P301L mice, the R47H variant exerted no overt change on the DAM signature.
Single-nucleus RNA sequencing told a similar story, in that a population of microglia expressing DAM genes was overrepresented in R47H-TREM2 female P301L mice compared with mice expressing the common variant of the microglial receptor.
Gan concluded that the tau-related effects of the R47H TREM2 variant are not equivalent to those of TREM2 deficiency. At least in females. While TREM2 deficiency reportedly dampens the DAM signature in both male and female P301L mice, the R47H mutation ramped it up and shifted it toward a more pro-inflammatory, potentially damaging profile in females, she said. Gan attributed the worse memory loss in female R47H-TREM2s to this shift. She emphasized that this sexual dimorphism was not due to more tau pathology in female P301L mice, but to an altered microglial response to that burden.
The prevailing view in the field holds that R47H saps TREM2 function. Gan acknowledged that her data appear to go against that. Still, she said, the mutation might reduce TREM2’s response to Aβ, which is known to bind TREM2, yet have a different effect in the context of tau pathology alone.
When asked about the shifted microglial response to tau that Gan reported in female mice, Hansen hesitated. The gender difference could come down to timing, he noted. He reported that in his PS2APP TREM2 knockout model, females develop Aβ pathology sooner than males do, but ultimately the sexes even out. Gan’s observation of sex dimorphism—albeit in a tau only model—was made when the mice were 7 to 9 months old. Hansen agreed with Gan’s point that while TREM2 clearly mediates Aβ-instigated tau pathology, the receptor’s function in the context of tau pathology alone could be different.
Gan is exploring the source of this sexual dimorphism in microglial responses further. MicroRNAs are known to fine-tune gene expression—could they be at play here? By comparing microRNA expression profiles in microglia isolated from male and female mice, graduate student Lay Kodama noted a slew of differentially expressed microRNAs. Kodoma also compared the mRNA transcriptomes of microglia isolated from wild-type and dicer knockout mice, which cannot generate mature microRNA. Removing dicer altered the expression of nearly 1,000 genes in male microglia, but only about 100 in female microglia, suggesting that microRNA disproportionately affect gene expression in microglia from males. Indeed, deleting dicer in male P301Ls exacerbated tau pathology, but in female P301Ls it did not. Overall, Gan interpreted her data to suggest that male and female microglia use different biological mechanisms to maintain homeostasis.
“These results indicate that males and females likely respond to AD-like neurodegeneration differently,” commented Li-Huei Tsai of Massachusetts Institute of Technology. “It would be very interesting to compare the transcriptomes of men and women who carry R47H in multiple cell types to see if there are differences,” she added.
Using single-cell RNA sequencing of human prefrontal cortex samples, Tsai recently reported sex differences in neuron and oligodendrocyte responses to AD, to which she attributed sex differences in AD risk and in white-matter pathology (Jul 2018 conference news; May 2019 news). Her study recovered too few microglia from the human samples to properly compare the sexes. However, at Keystone, Tsai reported that single-cell RNA sequencing from hippocampal samples, which have higher numbers of microglia, is underway, and could support such an analysis.
Next, Gan wants to test whether the sex differences are intrinsic to the cells or are imparted by the brain environment, as well as whether they hold true in human microglia.
Both these questions might find an answer with human microglia chimeric mouse models, such as the one developed by Matthew Blurton-Jones at the University of California, Irvine. At Keystone, Blurton-Jones characterized his model, in which immunodeficient mice receive transplants of iPSC-derived human microglial progenitors. Blurton-Jones generated them in response to mounting data showing that human microglia are different from their mouse counterparts in ways that matter greatly to the study of neurodegenerative disease (May 2019 news).
Alzforum covered some of this work at the AD/PD conference in Lisbon, along with findings from a similar chimeric model developed by Bart De Strooper at the U.K. Dementia Research Institute and KU Leuven (Apr 2019 conference news). Blurton-Jones had reported that when the recipient mice were 5xFAD, transplanted human microglia surrounded plaques but expressed different genes than the DAM signature of mouse microglia. At Keystone, he added that when the human microglia expressed a copy of R47H-TREM2, they were less energetic at rallying around plaques. Blurton-Jones has started transplanting microglia into P301L mice, and his initial findings suggest that transplanted human microglia nibble away at tau-laden neurons.
Both Gan and Blurton-Jones intend to use chimeric models to find out what is behind the sex differences that Gan saw in microglial responses to tauopathy. For example, by transplanting iPSC-derived microglia from women into male mice, or from men into female mice, the researchers can perhaps distinguish relative contributions of the brain environs from microglia-intrinsic properties that might drive the sex differences.
A smattering of other presentations at Keystone highlighted sex-specific differences in how microglia respond to a variety of perturbations. For example, Sandra Siegert from the Institute of Science and Technology in Klosterneuburg, Austria, reported that, in response to treatment with anesthetics, female microglia transformed into phagocytic cells that attacked structures surrounding parvalbumin-positive interneurons. Male microglia ultimately did the same, but responded more slowly. The findings suggest that female microglia may act faster on inhibitory interneurons to counteract dips in neuronal activity.
Julia Kuhn, who was a postdoc at the University of California, San Francisco, and is now at Genentech, reported a finding on pain. In response to peripheral nerve injury, Kuhn found, sensory neurons in the spinal cord pump out CSF-1, which signals through CSF-1R on spinal microglia (Guan et al., 2016). Males, but not females, are known to become hypersensitive to pain following such injuries. At Keystone, Kuhn reported why: The rush of CSF-1 in the spinal cord recruits T cells, which dampen microglial activation. In males, no T cells are recruited, and microglia become overactivated, stoking pain.
YajingXu of University College London reported that differences in the way males and females respond to pain could come down to how microglia sculpt nerve fibers during development. During the first week of life, microglia from both sexes engulfed portions of nerve fibers that receive touch and pain signals in the dorsal horn of the spinal cord, Xu found. When Xu nicked the hind paw of the mice during this time period, microglia responded by crowding the nerve fibers that transmitted the injury signal. However, only male microglia stepped up their engulfment of nerve fibers, essentially hijacking this normal developmental process, Xu said. She is investigating whether these differences in microglial engulfment of nerve fibers shape pain sensitivity in adulthood.
Overall, researchers agreed that microglia respond differently between the sexes in some instances, but what drives those differential responses, and how they ultimately affect the course of disease, remain to be explored.—Jessica Shugart
Dopaminergic Neurons Conjured from Astrocytes Restore Motion
Astrocytes are devoted nurturers of neurons—facilitating synaptic transmission, maintaining the blood-brain barrier, and repairing injuries are but a few of their ministrations. As if that weren’t enough, scientists described an instance where astrocytes made the ultimate sacrifice: by leaving their identity behind and becoming something else entirely. At the joint symposia Neurodegenerative Diseases: New Insights and Therapeutic Opportunities, and Neural Environment in Disease: Glial Responses and Neuroinflammation, held June 16–21 in Keystone, Colorado, Don Cleveland of the University of California, San Diego, reported on an experimental protocol whereby dialing down expression of a single gene transformed astrocytes into dopaminergic neurons within the substantia nigra of the mouse brain. No infusion of cells needed.
The in situ neuronal converts pumped out dopamine, and rescued motor deficits in a model of Parkinson’s disease, Cleveland reported. Researchers at the conference peppered Cleveland with questions and potential pitfalls of this approach. Perhaps only by converting to neurons could astrocytes—forever the wallflowers of brain research—spark this much excitement at a neurodegeneration meeting.
In fact, Cleveland kicked off his Keystone talk by extolling the virtues of antisense oligonucleotide therapy, which has resulted in an approved drug for spinal muscular atrophy, and is in various stages of clinical development to take down a cadre of disease-related proteins including huntingtin, tau, and more. But Cleveland suggested ASOs could be put to use to accomplish a different feat. Rather than knock down this or that disease protein, ASOs could give rise to brand-new neurons in the diseased, aging brain.
Researchers led by Fred Gage of the Salk Institute in La Jolla, California, previously developed protocols that could convert fibroblasts directly into neurons in a dish by adding in the right mix of transcription factors (Jun 2011 news). Across the street at UCSD, Cleveland’s colleague Xiang-Dong Fu subsequently simplified the conversion process. He found that turning down expression of a single protein—pyrimidine-tract-binding (PTB)—triggered fibroblasts to transform into bona fide neurons that sprouted axons and fired action potentials (Jan 2013 news on Xue et al., 2013; for review, see Hu et al., 2018). PTB is an RNA-binding protein. At Keystone, Cleveland presented the results of his collaboration with Fu, which took the logical, if ambitious, next step of sparking the conversion of astrocytes into neurons within the brain.
To pull off this form of “identity theft,” as Cleveland called it, the researchers targeted PTB with an ASO. In culture, this oligonucleotide induced the differentiation of mouse or human astrocytes into cells that expressed typical neuronal markers, and that differentiated into different kinds of neurons. A small proportion of the converted neurons expressed tyrosine hydroxylase (Th), a marker of dopaminergic neurons.
Could they orchestrate this conversion within the brain? To find out, the researchers injected wild-type mice with an adeno-associated virus (AAV) expressing a small-hairpin RNA (shRNA) to inhibit PTB expression, and a red fluorescent protein to mark infected cells. Fu lab postdoc Hao Qian and colleagues’ expression system glowed red in infected astrocytes, and remained red even if the cells converted into neurons. Cleveland reported at Keystone that one month after injection, 80 percent of red cells expressed neuronal, not astrocyte, markers. A quarter of the converted cells also expressed Th, suggesting they could make dopamine. In animals injected with a control virus expressing a nonspecific shRNA along with the red fluorescent protein, the marked cells remained astrocytes.
Next, the scientists tested the strategy in a mouse model of PD, in which they obliterated dopaminergic neurons on one side of the substantia nigra by injecting the neurotoxin 6-OHDA. Following ablation, the researchers injected the PTB-lowering or control virus directly into the substantia nigra. A month later, they found that while a neuronal desert surrounded the toxic lesion in mice receiving the control, converted neurons abounded in those with the PTB-lowering virus. The treatment raised neuronal numbers up to a third of their original levels. The 6-OHDA lesion docked dopamine levels in the nearby striatum, which receives dopamine from nigral neurons, by 80 percent. Treatment with the PTB-lowering virus restored dopamine levels back up to 66 percent of normal.
Would this be sufficient to stop the motor problems caused by the neurotoxic lesion? Cleveland reported that the ablation caused mice to walk in circles, reflecting loss of motor control on one side. However, animals treated with the PTB-lowering virus stopped circling two to three months later, while those receiving a control virus didn’t. Notably, the benefits of the PTB-lowering virus lasted up to one and a half years, while animals treated with the control virus still displayed the circling behavior.
Trying to link this improvement to induced neurons, the researchers next added another gene to their PTB-lowering virus. They loaded on a mutagenized version of a muscarinic acetylcholine receptor that deactivates neurons when it encounters the synthetic peptide CNO. Essentially, this addition allowed the researchers to deactivate converted neurons at will. Cleveland reported that while the PTB-lowering/mACh virus rescued the circling phenotype in lesioned mice, they relapsed back into circling when the researchers deactivated the converted neurons with CNO. The mice recovered mobility a second time after the injected CNO wore off. In all, the findings suggested that the conversion of astrocytes into neurons not only restored dopaminergic function in the 6-OHDA-poisoned mice, but also provided a durable rescue of motor deficits caused by the ablation.
Excitement was palpable in the room as researchers barraged Cleveland with questions. Charles Meshul of Oregon Health Sciences University in Portland asked whether the converted neurons formed functional circuits with the striatum. Cleveland said that while patch-clamp experiments suggest the neurons are functional and dopamine levels suggest they pump out the neurotransmitter, it is unclear whether they form connections akin to those of native nigral neurons. Fu later told Alzforum that they are addressing this question by measuring newly formed synapses between the converted neurons and existing circuits in the brain. Martin Kampmann of the University of California, San Francisco, wondered what triggered the astrocytes to become dopaminergic neurons, instead of some other type of neuron. Cleveland speculated that different astrocytic subtypes might be inclined to transform into distinct neuronal subsets upon PTB inhibition, but local cues within the nigra, where the virus was injected, likely also steered the converts into the dopaminergic type.
Others wondered about potential downsides of trading astrocytes for neurons. Cleveland acknowledged that he had yet to investigate consequences of lowering the astrocyte pool, and noted that any company developing ASOs against PTB should run dosing studies to evaluate side effects.
Others questioned how the strategy would work in the context of true PD, as opposed to an acute neurotoxic lesion. Cleveland said that remains to be seen. More work needs to be done in genetic models of the disease, he said. Later, he told Alzforum that in PD patients, the neuronal converts would have to withstand an ongoing neurodegenerative environment. Therefore, he sees astrocyte conversion working best in combination with other treatments that target the processes driving neurodegeneration.
Could this strategy assuage neuronal loss in other diseases? Cleveland said possibly, yes, although a targeted approach of replacing lost dopaminergic neurons in the substantia nigra has the highest chance of success. AaronGitler of Stanford University was impressed by Fu and Cleveland’s findings. He agreed that neuronal replacement could theoretically work for PD, but would be hard-pressed to bring back functional motor neurons lost in ALS. Motor neurons must sprout axons that form connections over vast distances, and Cleveland agreed that might be asking too much of a newly minted neuron.—Jessica Shugart
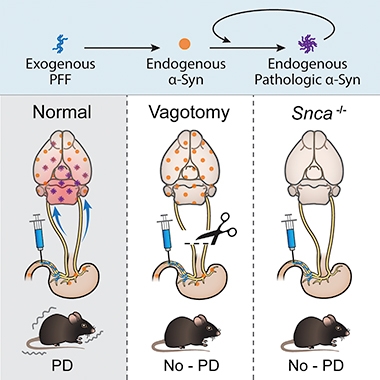
In PD Model, α-Synuclein Spreads from Intestine to Brain
The hypothesis that Parkinson’s disease could arise within the belly, not the brain, now gains support with a new mouse model. Valina Dawson of Johns Hopkins University, Baltimore, reported at a joint symposium held June 16-21 in Keystone, Colorado, that α-synuclein fibrils injected into the gut seeded the misfolding of normal mouse α-synuclein there. Over the course of 10 months, the aggregates spread into the midbrain and ultimately the cortex, where they killed neurons and triggered motor and cognitive deficits. Snipping the vagus nerve protected the brain. The study, co-led by Hopkins’ Ted Dawson and Han Seok Ko, was published in Neuron on June 17.
The gut hypothesis of Parkinson’s disease emerged in 2003 when neuroanatomists Heiko Braak and Kelly Del Tredici, both at the University of Ulm in Germany, spotted α-synuclein-laden inclusions within the enteric nervous system of people who had died with PD (Braak et al., 2003, and Jul 2011 news series). They proposed a staging scheme in which α-synuclein pathology spread from the gut to the vagus nerve then into the brain stem, midbrain, and ultimately to higher brain regions. However, the intestinal origins of PD have been difficult to prove. Dawson told the audience at Keystone that people who died with Lewy bodies that had not yet spread beyond the vagus nerve would not have been diagnosed with PD during life.
At Keystone, Dawson described how the new mouse model proves that the intestinal origin is plausible—at least in mice. Co-first authors Sangjune Kim and Seung-Hwan Kwon injected preformed fibrils (PFFs) of mouse α-synuclein into muscle layers of the duodenum and pylorus, which connects this first part of the small intestine to the stomach. These regions are heavily innervated by the vagus nerve. One month later, the researchers detected mouse pSer129-α-synuclein (p-syn) within the pylorus and duodenum, as well as in the medulla oblongata of the brain, where the vagus nerve cell bodies reside. By three months, p-syn had cropped up in the amygdala, and had started to accumulate in the substantia nigra, hypothalamus, and prefrontal cortex. By seven months, traces of the pathology had spread to the hippocampus, striatum, and olfactory bulb. By 10 months, p-syn aggregates filled the hippocampus, prefrontal cortex, olfactory bulb, and striatum, but had diminished in the amygdala, ventral midbrain, and medulla oblongata (see image below).
Propagation from the Gut. One, three, seven, and 10 months after PFFs were injected into the gut, p-syn aggregates (red dots) turned up in the dorsal motor nucleus of the vagus nerve. (DMV). They also were identified in the locus coeruleus, substantia nigra pars compacta, substantia nigra reticulate, basolateral amygdala, hippocampus, hypothalamus, cortex, prefrontal cortex, and striatum. MO is the medulla oblongata and OB is the olfactory bulb. [Courtesy of Kim et al., Neuron, 2019.]
This pathological cascade did not occur when fibrils were injected into α-synuclein knockout animals, or if α-synuclein monomers or PFFs of human α-synuclein were used. Together, these data suggested that the injected mouse α-synuclein PFFs seeded the misfolding of endogenous mouse α-synuclein in the gut, which propagated in a retrograde fashion into and throughout the brain. Snipping the vagus nerve shortly after the PFF injection—effectively cutting off the access route of the pathology into the brain—also nipped the pathology in the bud (see image below).
How did this spread affect the brain? The number of dopaminergic neurons in the substantia nigra took a nosedive by seven months after the injection, and by 10 months, more than half of those neurons had died. Dopamine levels in the brain started to drop three months after injection, and continued to plummet thereafter. Compared with control mice, the PFF-injected animals took longer to climb down a pole, fell more quickly off a spinning rod, and had weaker grip strength seven months after injection. They also dawdled over nest building, poorly remembered the location of a hidden platform, and failed to distinguish between novel and familiar objects. Again, α-synuclein knockout mice injected with PFFs had no neurodegeneration or behavioral deficits.
PD from the Gut? Injection of α-syn PFFs into the gut triggered the propagation of α-synuclein pathology into the brain (left), where it caused neurodegeneration. Vagotomy (middle) prevented the propagation and α-synuclein knockout mice were unaffected. [Courtesy of Kim et al., Neuron, 2019.]
Dawson pointed out that several other labs have tried, and failed, to spark PD-like pathology in the brain by targeting the mouse gut. She attributed her lab’s success to a number of factors, including their use of small fibrils, the hefty dose they injected (25 ug), their highly targeted injection adjacent to the vagus nerve, and how long they were willing to wait for the disease to unfold.
Aaron Gitler of Stanford University said that if it can be reproduced, the model could provide a useful way to study α-synuclein propagation from the gut. He said that the dependence on endogenous mouse α-synuclein suggests that the model is meaningful. Subhojit Roy of the University of Wisconsin in Madison agreed that the phenomenon was remarkable, but added that Dawson’s model still falls prey to the same artificiality that dogs all PFF injection models. “You do not see extracellular α-synuclein floating around in people with PD, so it is difficult to know how models like this relate to disease,” he said. “However the fact that extracellular α-synuclein can induce rapid intracellular aggregation is probably telling us something important.” Don Cleveland of the University of California, San Diego, wondered whether the pathological spread would be mitigated in mice expressing only a single copy of α-synuclein. Dawson has yet to test this.
In people, what would set off α-synuclein misfolding in the gut in the first place? Dawson suggested that problems with the gut microflora could somehow spark the process. There is evidence of an altered microbiome and inflammatory responses within the guts of PD patients (Dec 2016 news and May 2019 conference news).
Dawson also noted that levels of Poly ADP-ribose (PAR) are high in the gut. She previously implicated this branched chain ribose in aggregating and propagating α-synuclein in the brain (Kam et al., 2018).—Jessica Shugart
Do Microglia Finish Off Stressed Neurons Before Their Time?
Microglia not only gobble up detritus from dead cells, they also have a penchant for chowing down on live ones. At a Keystone joint symposia—Neurodegenerative Diseases: New Insights and Therapeutic Opportunities and Neural Environment in Disease: Glial Responses and Neuroinflammation—held June 16–21 in Keystone, Colorado, researchers described how microglia use different receptors to sense neurons that are stressed, dying, or behaving oddly. Microglia start eating their synapses, or even the entire cell. Sometimes the purging seems justified, scientists reported, for example when microglia use the TAM receptors Axl and Mer to clean up apoptotic debris, plaques, and overactive neurons. Other times, microglia take it too far, killing off functional neurons and synapses and impairing cognition. Researchers proposed strategies to block these overzealous microglia, which run amok when neurons are most stressed.
Microglial pruning of neurons is crucial during development, when these cells sculpt fledgling synaptic circuits. Their appetite for neurons is helpful during infections or injury, when they dispatch infected neurons or mop up apoptotic debris. However, if the immune cells nibble neurons that are merely stressed, they may contribute to neurodegeneration.
For example, scientists had previously reported that P2Y6—a purinoreceptor primarily known for its role in neuropathic pain—recognizes uridine 5'diphosphate (UDP), which is released from injured or stressed neurons (Koizumi et al., 2007). By blocking P2Y6 with the small molecule antagonist MRS2578, they spared neurons from an untimely death following intracerebral injection of lipopolysaccharide into rats. In culture, MRS2578 protected neurons exposed to Aβ from being eaten by overzealous microglia (Neher et al., 2014). At Keystone, Guy Brown of the University of Cambridge, U.K., extended this line of investigation to models of neurodegeneration and aging. He found that PY26 deficiency spared mice from neuronal loss and memory deficits induced by injection of Aβ oligomers, by tau pathology, or by the aging process itself.
YajingXu, University College London, wondered how sparing dying neurons from microglial engulfment could be beneficial, given that those neurons would remain dysfunctional or perhaps die soon. Brown replied that neurons releasing UDP might only be stressed, and not destined to die at that time. Similarly, Youtong Huang of the Salk Institute, San Diego, questioned whether deleting or blocking P2Y6 might lead to a build-up of apoptotic cells. Brown said that he has not checked for an accumulation of apoptotic debris in his models, but hypothesized that P2Y6 primarily facilitates microglial engulfment of viable cells—a process dubbed “phagoptosis” (for review, see Brown and Neher, 2012).
In her talk, Huang, a graduate student in Greg Lemke’s lab at Salk, reported how microglia use another set of receptors to keep the brain clear of apoptotic detritus in AD models. Lemke previously discovered that Tryo3, Axl, and Mer, collectively known as the TAM receptors, enable microglial engulfment of sick or dying neurons (see Fourgeaud et al., 2016, for review; Lemke 2017). From the microglial surface, the receptors recognize phosphatidylserine, a membrane phospholipid that flips from the inner to outer membrane when neurons are stressed or apoptotic. Preventing phosphatidylserine exposure on the cell surface protected virally infected neurons from being culled by microglia (Tufail et al., 2017). Other researchers have found TAM receptors on the surface of myeloid cells surrounding plaques in AD mouse models (Feb 2015 news).
At Keystone, Huang reported that in 9.5-month-old APP/PS1 mice, microglia surrounding plaques expressed gobs of Axl and Mer. While these cells always make some Mer, only activated microglia turn on Axl. Huang said that microglia mingling with plaques had 30-fold higher levels of Axl than those farther away, in keeping with the 25-fold upregulation of Axl previously reported in disease-associated microglia (DAM). Huang showed additional data implying, essentially, that microglia expressing Axl/Mer, their ligands, phosphatyldserine, and Aβ all mingle together around dystrophic neurites.
Huang believes that microglial TAM receptor expression is an attempt to limit AD pathogenesis. In agreement with this idea, she found that the survival of APP/PS1 mice plummeted when Axl and Mer were knocked out. Forty percent of the animals died of seizures in their first year of life. Seizures occur in AD. The surviving 1-year-old APP/PS1 mice lacking Axl and Mer had worse memory loss on a contextual fear conditioning test than APP/PS1 controls. Huang also observed a build-up of cleaved caspase-3 puncta in the hippocampus, indicative of uncleared apoptotic cells.
Brown noted that in addition to marking apoptotic cells, phosphatidylserine also decorates the cell surface in response to increases in neuronal activity. He wondered whether a failure to clear those hyperactive neurons in Axl/Mer knockout mice could lead to a buildup of seizure-provoking neurons, perhaps explaining why the animals tend to die young from seizures. Huang said the lab is investigating the idea, as well as the role of TAM receptors in clearance of Aβ plaques.
Beth Stevens of Children’s Hospital in Boston discussed another form of microglial regulation. Stevens previously reported that microglial pruning of synapses, a normal developmental process, switches into overdrive in AD models (Apr 2016 news; Shi et al., 2017). Others have spotted overzealous pruning in tauopathy mice (Jul 2018 news). Essentially, microglia target synapses adorned with complement proteins.
At Keystone, Stevens asked what triggers synapses to start dressing up in complement in the first place. She turned to models of Huntington’s disease to address this question, because neurons in the cortico-striatal circuit are particularly vulnerable to the disease. Using two mouse models—BACHD and Hdh-Q175—postdoc Daniel Wilton found a disconnection in the pre- and postsynaptic compartments in this circuit starting at 3 months of age. Complement proteins crowded the vulnerable circuits, and microglia in the region were loaded with neuronal synapses. Treating mice with an antibody that blocks C1q not only spared synapses but, according to preliminary data, it also restored functional synaptic firing in the cortico-striatal circuit. Some of this work was previously presented at the 2015 Society for Neuroscience annual meeting (Nov 2015 conference news).
Would saving these synapses help? Indeed, Stevens reported that in preliminary experiments done in collaboration with William Yang at the University of California, Los Angeles, 12-month-old C3-deficient BACHD mice managed to balance on a spinning rod better than their C3-expressing counterparts, though they were not quite as agile as wild-type. Similar to BACHD mice treated with a C1q antibody, C3 deficiency spared synapses in the cortico-striatal circuit.
What about the human disease? Using postmortem brain tissue from people at different stages, Stevens identified progressive synapse loss, as well as an uptick in complement proteins, in the striatum. Notably, this synapse loss was apparent even in people who had died in the early stages of HD. Stevens also reported preliminary CSF findings from two HD patient cohorts—HD Clarity and Enroll HD—in which some complement proteins, C3 and iC3b in particular, rose in CSF but not plasma during presymptomatic stages. That said, CSF complement levels overlapped significantly between the different disease stage groups.
Stevens stopped short of addressing exactly what causes the vulnerable neurons in the cortico-striatal circuit to wave the complement flag, but speculated that aberrant neuronal activity and phosphatidylserine could be involved.
In neurodegenerative disease, malfunctions in neuronal circuitry could be an early trigger of damaging microglial responses, Stevens said. In his talk, Paul Worley, Johns Hopkins University in Baltimore, described how the immediate early genes Arc and Neuronal Pentraxin 2 (NPTX2) might facilitate those initial changes to neuronal circuitry, at least in Alzheimer’s disease. Worley reviewed his body of published work on the two proteins, which are important in memory consolidation and maintaining the brain’s circuitry. In a nutshell, Arc is rapidly translated in dendrites, and serves to weaken inactive synapses, while NPTX2 quells hyperexcitability in pyramidal neurons by strengthening their connections with inhibitory interneurons (Xiao et al., 2017).
Worley proposed that NPTX2 could be required to protect neurons from hyperactivity caused by Aβ or other insults. Interestingly, Worley and colleagues reported that CSF levels of NPTX2 drop as AD worsens, and correlate with loss of functional connectivity, suggesting this synaptic regulator could make a useful biomarker for AD progression (see Soldan et al., 2019). At the Alzheimer’s Association International Conference, which just concluded in Los Angeles on July 18, several proteomic and fluid-based studies noted a decline of NPTX2 in the run-up to AD dementia, as well (Aug 2019 coference news).
Stevens noted that the early circuitry imbalances described by Worley are likely what instigates microglia hunger pangs early in neurodegenerative disease.—Jessica Shugart




Comments
No Available Comments
Make a Comment
To make a comment you must login or register.