Changing With the Times: Disease Stage Alters TREM2 Effect on Tau
Quick Links
Variants in the TREM2 gene increase the chances of getting Alzheimer’s disease by two- to fourfold. Scientists have done much digging to figure out how this transmembrane receptor influences microglial responses to Aβ plaques. Now, two groups report how it influences the second major hallmark of AD—tau. One, led by David Holtzman, Washington University School of Medicine in St. Louis, reports in the October 9 Proceedings of the National Academy of Sciences that TREM2 accelerates neurodegeneration in a pure tauopathy model. Another group, led by Bruce Lamb, Indiana University School of Medicine, Indianapolis, describes in an upcoming paper in Molecular Neurodegeneration how TREM2 plays a protective role in early disease by tamping down tau hyperphosphorylation and aggregation. Together they suggest that TREM2 assumes different roles at early versus late stages of disease.
- In one mouse model of tauopathy, TREM2 worsens neurodegeneration.
- In another, TREM2 puts a lid on pathology.
- Microglia appear to evolve as disease progresses.
“It’s exciting that TREM2 deficiency would yield apparently different results, because it shows that microglia fine-tune reactions to their environmental signals,” said Monica Carson, University of California, Riverside. Terrence Town, University of Southern California, Los Angeles, noted that both studies were incredibly internally consistent. Combined, the results represent TREM2’s contrasting responses to two different forms of tau, he said. “One looks at the physiological interplay between TREM2 and tau, while the other the effect of TREM2 on tau pathology."
A third paper, from the lab of Li-Huei Tsai at the Massachusetts Institute of Technology, adds to the idea that microglial phenotypes morph as disease progresses. In the October 10 Cell her group reports that mouse microglia divide and proliferate early in neurodegenerative disease, then unleash a heterogeneous mix of immune responses.
Holtzman and co-first authors Cheryl Leyns and Jason Ulrich crossed TREM2 knockouts with the PS19 mouse, which expresses the P301S mutant form of human tau that causes familial frontotemporal dementia. By nine months, PS19 mice aggressively accumulate tau deposits, have extensive gliosis, and lose vast numbers of neurons. To their surprise, Leyns and colleagues found that at nine months, the PS19/TREM2 knockouts had substantially less atrophy in their brains than PS19 mice had, as judged by little ventricular enlargement, more cortical volume, and less synaptic loss (see image below). Interestingly, the amount of phosphorylated and aggregated tau did not differ between the crosses and control PS19 mice. At the same time, the PS19/TREM2 knockout mice had much less microgliosis and astrogliosis in the hippocampus and piriform cortex than PS19 controls, and their microglia expressed fewer markers of activation and inflammation.
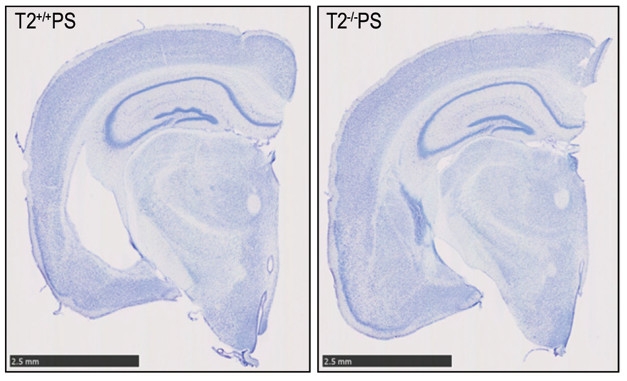
TREM2 Worsens Neurodegeneration. Brain slices of PS19 mice reveal less neuronal staining (blue), and therefore more neurodegeneration, with normal TREM2 (left) compared with TREM2 knockouts (right). [Image courtesy of Cheryl Leyns, PNAS.]
How does this data fit with TREM2 loss-of-function variants being major risk factors for dementia? The authors wrote that TREM2 could be helpful when it comes to Aβ plaques, containing them and protecting neighboring neurites (Wang et al., 2016; Yuan et al., 2016). However, when it comes to dealing with tau tangles, TREM2 appears to be detrimental, ramping up both inflammation and neurodegeneration. Holtzman’s group is now repeating this experiment with a mouse model that has both Aβ and tau pathologies to see how the lack of TREM2 affects the two together.
Holtzman points out that while these results are from mice, they may have therapeutic implications. “This is the first paper we’re aware of to say that the absence of TREM2 is associated with less neurodegeneration in Alzheimer’s disease,” he told Alzforum. This may give pause to pharmaceutical companies trying to come up with ways to target TREM2 as a therapeutic strategy. “The conventional wisdom from previous data is you might want to activate TREM2; this suggests you’d want to inhibit it, at least in the neurodegenerative phase of AD,” said Holtzman.
As described in the second paper, Lamb, first author Shane Bemiller, and colleagues crossed TREM2 knockouts with the htau mouse model that carries normal human tau under control of the human promoter in place of the murine endogenous gene. In this model, tau becomes hyperphosphorylated at around three months of age and starts to aggregate at around six months. The researchers found much more hyperphosphorylated and aggregated tau in the cortical layers of six-month-old crosses than in those of age-matched htau mice. Microglia in the cortex and hippocampus were smaller in the crosses and they had thinner processes with fewer branches. In addition, dramatic increases in JNK, GSK3β, p38, and ERK kinase signaling—all involved in tau phosphorylation—started at three months.
The results imply that TREM2 normally protects against tau pathology, microglial activation, and stress signaling, at least in the early stages of tau pathology modeled by these mice, said Lamb. Holtzman’s model likely represents a later stage of disease marked by neurodegeneration and inflammation, when TREM2 might accelerate demise, he said. “TREM2 may be playing different roles at different stages of disease progression.” Bemiller will examine these mice further as they age and see how TREM2 affects ongoing pathology. “I think it is critical to gather data throughout the aging process and development of pathology in these models,” added Lamb.
Oleg Butovsky, Brigham and Women’s Hospital, Boston, agreed that TREM2 could have different roles depending on disease stage and model. “In one model TREM2 could be protective, in a second it could be detrimental; we don’t know whether TREM2 is always going to be good or bad,” he said. He agreed with Holtzman that researchers will have to be careful about whether they take agonistic or antagonistic approaches to TREM2 in clinical applications. “It should be carefully weighted on disease and stage of disease.”
Tsai’s data support the idea that microglia tailor their response to a changing environment and are heterogeneous in the face of progressing neurodegeneration. First author Hansruedi Mathys and colleagues used single-cell RNA sequencing to track the transcriptome of p25 mice just before, as well as one, two, and six weeks after inducing neurodegeneration. In this mouse, developed by the Tsai lab and used mostly by this group, turning on p25 activates Cdk5 kinase, ultimately leading to excess Aβ production, tau phosphorylation, and severe neurodegeneration (Cruz et al., 2003).
One week after induction of p25, mouse microglia started to pump out genes associated with mitosis, implying they were rapidly dividing. Over the next five weeks, they transitioned from this proliferative state to one marked by upregulation of immune response genes. These microglia came in two distinct flavors—some produced MHCII genes and were spread diffusely through the brain, others pumped out interferon genes typical of an antiviral response and were clustered around neuronal cell bodies.
It is unclear how these late-response microglia differ in function, said Tsai. They share more than two-thirds of their upregulated genes with disease-associated microglia (DAM), which were reported to surround plaques in six-month-old 5xFAD mice and depended on TREM2 (Jun 2017 news on Keren-Shaul et al., 2017). However, many interferon and antiviral genes upregulated in late-response microglia were not observed in DAM, suggesting they are not exactly the same, Tsai said. Tsai believes the early response microglia she observed precede any DAM stage previously reported. She plans to look in postmortem human brains to see if microglia undergo a similar evolution of microglial activation states in disease.—Gwyneth Dickey Zakaib
References
Research Models Citations
News Citations
Paper Citations
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM, Colonna M. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med. 2016 May 2;213(5):667-75. Epub 2016 Apr 18 PubMed.
- Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 2016 May 18;90(4):724-39. PubMed.
- Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH. Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron. 2003 Oct 30;40(3):471-83. PubMed.
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M, Amit I. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell. 2017 Jun 15;169(7):1276-1290.e17. Epub 2017 Jun 8 PubMed.
Other Citations
Further Reading
Papers
- Leyns CE, Holtzman DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener. 2017 Jun 29;12(1):50. PubMed.
- Kim SM, Mun BR, Lee SJ, Joh Y, Lee HY, Ji KY, Choi HR, Lee EH, Kim EM, Jang JH, Song HW, Mook-Jung I, Choi WS, Kang HS. TREM2 promotes Aβ phagocytosis by upregulating C/EBPα-dependent CD36 expression in microglia. Sci Rep. 2017 Sep 11;7(1):11118. PubMed.
- Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, Loginicheva E, Gilfillan S, Cella M, Virgin HW, Unanue ER, Wang Y, Artyomov MN, Holtzman DM, Colonna M. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease. Cell. 2017 Aug 10;170(4):649-663.e13. PubMed.
- Efthymiou AG, Goate AM. Late onset Alzheimer's disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017 May 26;12(1):43. PubMed.
Primary Papers
- Leyns CE, Ulrich JD, Finn MB, Stewart FR, Koscal LJ, Remolina Serrano J, Robinson GO, Anderson E, Colonna M, Holtzman DM. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A. 2017 Oct 24;114(43):11524-11529. Epub 2017 Oct 9 PubMed.
- Mathys H, Adaikkan C, Gao F, Young JZ, Manet E, Hemberg M, De Jager PL, Ransohoff RM, Regev A, Tsai LH. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017 Oct 10;21(2):366-380. PubMed.
- Bemiller SM, McCray TJ, Allan K, Formica SV, Xu G, Wilson G, Kokiko-Cochran ON, Crish SD, Lasagna-Reeves CA, Ransohoff RM, Landreth GE, Lamb BT. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol Neurodegener. 2017 Oct 16;12(1):74. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Cambridge
The papers by the Holtzman and Lamb groups describe how TREM2 plays a protective role in early disease by tamping down tau hyperphosphorylation and aggregation. The results imply that TREM2 normally protects against tau pathology.
We would like to add our findings. We reported that TREM2 plays a critical role in inflammation and is essential for neuroplasticity and myelination in an AD transgenic mouse model. We described the presence of TREM2 in oligodendrocytes, in white-matter tracts of the olfactory bulb, corpus callosum, and striatal bundles, indicating that TREM2 protein may have a role in myelination. This would imply that the lack of functioning TREM2 could be a contributing mechanism in myelination deficits seen in neurodegenerative diseases and could increase the susceptibility for neuronal loss. We have shown by primary neuronal and glial culture that TREM2 protein was present in all cells (especially in hippocampal neurons and oligodendrocytes), indicating that TREM2 could be involved in neuroplasticity and neuroprotection (Raha et al., 2016).
Using human brain sections from AD and DS we provide evidence, for the first time, that soluble TREM2 originates in bone marrow, is transported to the brain by a subset of macrophages before being taken up by microglia in Down’s syndrome subjects (Raha-Chowdhury et al., 2017, JAD in press).
The TREM2 (R47H) mutation, which has been strongly linked with an increased risk of AD, was found in two DS subjects, and in both we observed gross morphological changes in megakaryocytes and erythrocytes. In erythrocytes, we also discovered impairment of TREM2 trafficking to the erythrocyte plasma membrane that could influence the amyloid clearance mechanism thought to be important in AD pathogenesis. TREM2 protein levels in brain and sera declined with age and disease progression in AD and DS.
Our findings support those observed by the Holtzman group that TREM2 could be neuroprotective, and understanding the molecular signaling pathways mediated by TREM2 may reveal novel therapeutic targets.
References:
Raha AA, Henderson JW, Stott SR, Vuono R, Foscarin S, Friedland RP, Zaman SH, Raha-Chowdhury R. Neuroprotective Effect of TREM-2 in Aging and Alzheimer's Disease Model. J Alzheimers Dis. 2017;55(1):199-217. PubMed.
Raha-Chowdhury R, Henderson JW, Raha AA, Stott SR, Vuono R, Foscarin S, Wilson L, Annus T, Fincham R, Allinson K, Devalia V, Friedland RP, Holland A, Zaman SH. Erythromyeloid-Derived TREM2: A Major Determinant of Alzheimer's Disease Pathology in Down Syndrome. J Alzheimers Dis. 2018;61(3):1143-1162. PubMed.
Make a Comment
To make a comment you must login or register.