Squelching ApoE in Astrocytes of Tau-Ravaged Mice Dampens Degeneration
Quick Links
Almost 30 years after ApoE was pegged as an Alzheimer’s risk gene, researchers are still investigating fundamental questions about how the apolipoprotein sways neurodegenerative disease processes. For one, which cell type is responsible for producing forms of the protein that beckon neurodegeneration? On April 7 in Neuron, researchers pinned some of the blame on astrocytes, which release the lion’s share of ApoE in the brain. Led by David Holtzman and Jason Ulrich at Washington University in St. Louis, the study found that in human ApoE knock-in mice, silencing astrocyte ApoE4, but not ApoE3, assuaged neurodegeneration instigated by tau pathology.
- Removal of ApoE4, but not E3, from astrocytes stemmed neurodegeneration in tangle-bearing mice.
- It also lessened tau pathology, synaptic pruning by microglia.
- Biggest beneficiaries: a subset of cortical neurons that amass RNA-binding proteins.
Turning off the astrocyte ApoE4 spigot just as tau started to accumulate not only stemmed subsequent brain shrinkage, it also dampened tau accumulation and profoundly influenced gene expression in multiple cell types, including neurons, oligodendrocytes, and microglia. Without made-in-astrocytes ApoE4, microglia were less prone to assume a hostile, neurodegenerative stance, and feasted less on fragile synapses. The findings suggest—at least in this mouse model of tauopathy—that astrocytic ApoE4 exacerbates most neurodegenerative phenotypes and likely does so via cross-talk with other cells.
“The paper highlights how the apolipoprotein, when produced in astrocytes, can exert effects in multiple cell types known to participate in pathogenic pathways of AD,” said Guojun Bu of the Mayo Clinic in Jacksonville, Florida. It also reiterates the potential gain of toxic function conferred on ApoE4 relative to ApoE3, he said. Bu, Holtzman, and other investigators agreed that the study supports the idea of dampening ApoE4 expression as a therapeutic strategy (Feb 2021 news).
While early research focused on how ApoE influences Aβ accumulation, more recently attention has shifted to tau. In a dramatic example of the strong tie between ApoE and tau, a woman carried an autosomal-dominant AD mutation and also happened to carry a rare protective variant in ApoE3 called the Christchurch mutation. She had a brain full of amyloid but hardly any tau tangles (Nov 2019 news).
Alas, if toxic forms of tau are around, ApoE makes things worse. Using human ApoE knock-in mice crossed to the P301S tau model of tauopathy, Holtzman and colleagues previously reported that expression of ApoE—in particular, ApoE4— exacerbated neurodegeneration (Sep 2017 news). Mice devoid of all ApoE fared best, with hardly any signs of disease. Subsequently, the scientists reported that microglia were required to dole out ApoE4’s damaging effects in tauopathy (Oct 2019 news). Microglia only express ApoE under pathological conditions, while astrocytes churn it out constitutively (Sep 2017 news). Hence, a key question remained unanswered: Did the ApoE4-microglia axis of neurodegeneration stem from microglial expression of ApoE4, or could ApoE4 made by astrocytes instigate the noxious microglial behavior?
To get at this question, first author Chao Wang and colleagues began what became a three-year saga of mouse husbandry. The researchers requisitioned Taconic to create custom knock-in mice that carry floxed, Cre-removable human ApoE3 or ApoE4 genes instead of their endogenous mouse ApoE gene. They crossed them to Aldh1l1-Cre/ERT2 BAC transgenic mice. These can be induced to express the Cre recombinase driven by the promoter for astrocyte-specific aldehyde dehydrogenase 1 family member L1. This shuts off ApoE expression in astrocytes (Srinivasan et al., 2016). Tamoxifen added to the drinking water flipped the Cre switch. The scientists then bred these animals with control or tau P301S mice, which develop tauopathy. Ultimately, this generated P301S Tau/Aldh1l1-Cre/ApoE3flox/flox and Tau/Aldh1l1-Cre/ApoE4flox/flox—TAFE3 mice and TAFE4 mice, for short. In them, scientists can turn off human ApoE selectively in astrocytes at any time.
So what happened? The researchers waited until the TAFE3 and 4 mice reached 5.5 months of age—just as tau pathology started to appear—and then switched off their astrocytic ApoE. This reduced the total amount of ApoE in the cortex, an area vulnerable to tau accumulation, by 50 to 80 percent. In the cerebellum, which is unaffected by tau pathology, ApoE plummeted by more than 90 percent. These numbers made sense given that under physiological conditions, astrocytes are the primary source of ApoE. An oil placebo dropped into the drinking water did not affect ApoE expression.

Sparing Shrinkage. In TAFE3 female mice, turning off ApoE3 expression in astrocytes with tamoxifen (TAM) did not slow brain atrophy; turning off ApoE4 in TAFE4 mice did lessen atrophy (far right). This effect was weaker in males. [Courtesy of Wang et al., 2021.]
By 9.5 months of age, all the mice, regardless of which ApoE isoform they expressed or whether it was turned on or off, had brain shrinkage by multiple measures. This atrophy was greatest in ApoE4 mice. Shutting off astrocytic ApoE expression had no effect on atrophy in E3-expressing mice, but reduced atrophy by about 25 percent in E4-expressing mice (see image above). These effects were more pronounced in females. Unlike control mice, those whose astrocytic ApoE4 was off kept busy building nests, an innate behavior. Taken together, this suggested that astrocytic ApoE4 exacerbates neurodegeneration in response to tau accumulation.
By that age, removing ApoE from astrocytes also influenced the extent of tau pathology. In females, depleting astrocytic ApoE3 or ApoE4 reduced levels of phospho-tau-181 detected in cortical extracts. For males, there was a trend toward reduction, but only upon removal of ApoE4. Using immunohistochemistry, the researchers detected a drop in phospho-tau, as gauged by reactivity with the AT8 antibody, in cortical slices of female, but not male, mice sans astrocytic ApoE4. Together, the findings suggested that astrocytic ApoE4 exacerbated tau deposition, particularly in females. That said, removing either form of ApoE from astrocytes alone was not sufficient to completely stop tau pathology in either sex.

Tamp Down Tau. Phospho-tau levels in TAFE4 mice (left) were reduced (right) by abolishing ApoE4 expression. [Courtesy of Wang et al., Neuron, 2021.]
How did cells across the tau-tangled brain react to ApoE’s ouster? To address this, the researchers turned to single-nucleus RNA sequencing. They used hippocampal tissue from 9.5-month-old TAFE3 or TAFE4 or control mice that had ingested oil or tamoxifen at 5.5 months of age. Based on the transcriptomes of more than 71,000 nuclei isolated in toto, the researchers identified 16 clusters of cells. These included 11 subsets of neurons, and one each of oligodendrocytes, oligodendrocyte progenitor cells, choroid plexus epithelial cells, astrocytes, and microglia.
Among the neuronal subsets, the researchers spotted two clusters—1 and 3—that plummeted in response to tau pathology. Interestingly, removal of ApoE from astrocytes protected against this tau-mediated cell loss, with ApoE4 removal having the strongest effect.
In contrast, another transcriptional clique of neurons—cluster 6—dramatically expanded in mice with tau pathology. Knocking down astrocytic ApoE3 or E4 curtailed this expansion. These neurons uniquely expressed a cadre of RNA-binding proteins—Arrp21 and R3hdm1 among them. They also expressed Rorb, a protein recently reported to mark subsets of neurons vulnerable to tau pathology (Jan 2021 news). The researchers believe this transcriptome might represent the neurons’ response to ongoing tau pathology.
Indeed, RNA-binding proteins have been shown to mingle with tau and coax its aggregation. In particular, Arpp21 has been spotted in stress granules in neurons, which have also been linked to the stabilization of toxic tau oligomers and accelerated neurodegeneration (Nov 2017 news; Rehfeld et al., 2018). Investigating a potential connection between the two proteins with immunohistochemistry, the researchers spotted Arrp21 and phospho-tau co-mingling in P301S mice expressing either isoform of ApoE. Astrocytic ApoE4, but not ApoE3, depletion dramatically reduced this association in neurons.
How astrocytic ApoE4 might influence tau’s association with RNA-binding proteins in neurons remains unclear, Holtzman said. Altered neuronal signaling, perhaps in response to worse damage in ApoE4 mice, could disrupt many neuronal functions.
Astrocytes themselves also responded to tau pathology, and their response in turn was modulated by extinguishing their ApoE. The scientists defined three subclusters based on transcriptional profiling. One expressed homeostatic genes, one was reactive, and a third appeared senescent. Removing ApoE—especially E4—enlarged the pool of homeostatic astrocytes, while shrinking the reactive one. Nicholas Seyfried of Emory University in Atlanta commented that these results align with proteomic studies on thousands of human brains. In those, astrocytes expressing a similar subset of genes as those seen in the mouse reactive subtype correlated with neuropathology (May 2020 news).
Not to be outdone, oligodendrocytes also shifted their transcriptomes depending on tau and ApoE. The myelin-producing glia ratcheted up expression of the complement protein C4b in the presence of tangles, and draining astrocytes of either isoform of ApoE dampened this pro-inflammatory effect. The findings mesh with recent studies that identified C4b-expressing oligodendrocytes in models of amyloidosis (Nugent et al., 2020; Zhou et al., 2020).
Finally, the researchers examined how astrocytic ApoE molded microglial responses to tau pathology. Within the larger transcriptomic cluster of microglia, they found three subclusters. One was enriched for genes expressed under conditions of homeostasis, a small subset appeared to be peripheral macrophages, and the third had a disease-associated microglia (DAM)/neurodegenerative (MGnD) signature (Jun 2017 news; Sep 2017 news).
As expected, this third, pathological subset was dramatically expanded in the presence of tau pathology. Nixing astrocytic ApoE did not change the relative proportions of the different subsets of microglia in tau-ridden mice. That said, using immunohistochemistry of hippocampal sections, the researchers did see that when ApoE4 was gone from astrocytes, more microglia expressed the homeostatic marker P2ry12 and downshifted expression of select DAM/MGnD genes, including MHC II, Spp1, and Clec7a. Curiously, astrocytic ApoE did not significantly influence microglial expression of ApoE, a DAM gene.
Yes, this is complex, but hang in there, dear reader. Next, the scientists asked if these transcriptomic shifts influenced how microglia behave—in particular, their appetite for synapses. Compared to control mice, tangle-ridden P301Ss had about 20 percent fewer synapses in their entorhinal and piriform cortices by 9.5 months of age, and microglia were spotted having stuffed their lysosomes with synaptic material. Crucially, removing ApoE4 from astrocytes at month 5.5 meant the number of synapses was near-normal four months later, and markedly reduced the amount of synaptic material in the bellies of the microglial beasts.
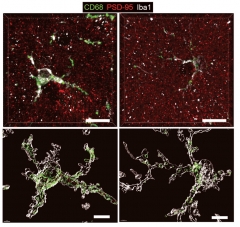
Appetite for Destruction. In TAFE4 mice (left), microglia expressed more lysosomal CD68 (green), and engulfed more synapses (PSD95, red) than did microglia in TAFE4 mice without astrocytic ApoE4 (right). [Courtesy of Wang et al., Neuron, 2021.]
In all, the findings suggest that ApoE4 from astrocytes somehow riles up damaging microglial responses to tau pathology, and this despite microglia expressing their own ApoE under these conditions. What is so special about ApoE from astrocytes? Holtzman pointed out that it carries more lipids than ApoE from microglia. That could influence the way the apolipoprotein interacts with microglial cell-surface receptors. He speculated that under conditions of neuronal stress, such as tauopathy, ApoE4, along with other local factors such as complement proteins, might somehow bait microglia to eat neuronal synapses.
The findings argue against a key role for microglial ApoE during pathology in this model, commented Jesse Hanson of Genentech in South San Francisco. “Therefore, the role of activated microglia in mediating tau-dependent pathology is likely not so much via production of ApoE, but rather as a mediator of neuronal damage in response to the harmful inflammatory state of the brain that is caused by tau pathology, and exacerbated by [astrocytic] ApoE4,” he wrote.
Ulrich and Holtzman don’t think their data entirely rule out a role for microglial ApoE4 in tau-mediated neurodegeneration. After all, shutting off ApoE4 in astrocytes had a subtler benefit than complete knockout of ApoE, which, as the researchers had previously reported, erased nearly all traces of disease in P301S mice. This implies that ApoE derived from non-astrocytic cell types still influences neurodegeneration.
“This study re-emphasizes, and expands on, previous work by the Holtzman lab and others. It makes very clear that astrocytic APOE4 represents a toxic gain of function,” wrote Priyanka Narayan of the National Institutes of Health in Bethesda, Maryland. “Given APOE’s role in lipid metabolism, and the involvement of lipids in nearly every cellular process, it is possible that lipid state is a key mediator of astrocytic APOE’s inter- and intracellular effects,” she added.
Overall, the paper illustrates the striking degree of cross-talk between different cell types that steers the course of neurodegeneration. “If you modulate ApoE in astrocytes, it affects neurons, oligodendrocytes, and microglia in different ways,” Seyfried told Alzforum. In other words, when astrocytic ApoE binds to surface receptors on different cell types, different downstream signaling events may be triggered in those cell types, Seyfried noted.
On the lipidation idea, he added that astrocytic ApoE may have a unique structure and lipid content compared to ApoE derived from other cell types. Holtzman noted another possibility. Perhaps a heightened reactivity of ApoE4-producing astrocytes influences microglia via inflammation-stoking proteins, not via ApoE itself.
Bu hopes that future studies will investigate what’s behind the sex differences reported in the paper. They add to recent work suggesting that microglia in males and females react differently to all manner of insults, and that deficiency of the microglial cell surface receptor TREM2 differentially alters male and female microglial responses to tau (Jun 2018 news; Jul 2019 conference news). ApoE4 is a stronger driver of AD risk in women than in men (Sep 2017 news).—Jessica Shugart
References
News Citations
- Would ApoE Make a Better Therapeutic Target Than Aβ?
- Can an ApoE Mutation Halt Alzheimer’s Disease?
- ApoE4 Makes All Things Tau Worse, From Beginning to End
- In Tauopathy, ApoE Destroys Neurons Via Microglia
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- Selective Vulnerability News: RORB Neurons Are First Victims of Tangles
- Stress Granule Protein Stabilizes Tau Oligomers, Hastens Neurodegeneration
- Massive Proteomics Studies Peg Glial Metabolism, Myelination, to AD
- Hot DAM: Specific Microglia Engulf Plaques
- Girl Power? In Mice, Female Microglia Protect Against Ischemic Injury
- Down to Sex? Boy and Girl Microglia Respond Differently
- New Look at Sex and ApoE4 Puts Women at Risk Earlier than Men
Research Models Citations
Paper Citations
- Srinivasan R, Lu TY, Chai H, Xu J, Huang BS, Golshani P, Coppola G, Khakh BS. New Transgenic Mouse Lines for Selectively Targeting Astrocytes and Studying Calcium Signals in Astrocyte Processes In Situ and In Vivo. Neuron. 2016 Dec 21;92(6):1181-1195. Epub 2016 Dec 8 PubMed.
- Rehfeld F, Maticzka D, Grosser S, Knauff P, Eravci M, Vida I, Backofen R, Wulczyn FG. The RNA-binding protein ARPP21 controls dendritic branching by functionally opposing the miRNA it hosts. Nat Commun. 2018 Mar 26;9(1):1235. PubMed.
- Nugent AA, Lin K, van Lengerich B, Lianoglou S, Przybyla L, Davis SS, Llapashtica C, Wang J, Kim DJ, Xia D, Lucas A, Baskaran S, Haddick PC, Lenser M, Earr TK, Shi J, Dugas JC, Andreone BJ, Logan T, Solanoy HO, Chen H, Srivastava A, Poda SB, Sanchez PE, Watts RJ, Sandmann T, Astarita G, Lewcock JW, Monroe KM, Di Paolo G. TREM2 Regulates Microglial Cholesterol Metabolism upon Chronic Phagocytic Challenge. Neuron. 2020 Mar 4;105(5):837-854.e9. Epub 2020 Jan 2 PubMed.
- Zhou Y, Song WM, Andhey PS, Swain A, Levy T, Miller KR, Poliani PL, Cominelli M, Grover S, Gilfillan S, Cella M, Ulland TK, Zaitsev K, Miyashita A, Ikeuchi T, Sainouchi M, Kakita A, Bennett DA, Schneider JA, Nichols MR, Beausoleil SA, Ulrich JD, Holtzman DM, Artyomov MN, Colonna M. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer's disease. Nat Med. 2020 Jan;26(1):131-142. Epub 2020 Jan 13 PubMed. Correction.
Further Reading
Primary Papers
- Wang C, Xiong M, Gratuze M, Bao X, Shi Y, Andhey PS, Manis M, Schroeder C, Yin Z, Madore C, Butovsky O, Artyomov M, Ulrich JD, Holtzman DM. Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron. 2021 May 19;109(10):1657-1674.e7. Epub 2021 Apr 7 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Mayo Clinic
This is a really important and definitive study demonstrating that astrocyte-derived ApoE has a major impact on tau accumulation and associated pathologies. Even though many labs are studying microglial ApoE in Alzheimer’s disease, the major source of ApoE in the brain is still astrocytes, and they have convincingly demonstrated that here, as in Fig. 1 where conditional genetic deletion in astrocytes dropped ApoE by more than 90 percent. This is not to say that microglia don’t make a lot of ApoE on a “per cell” basis, but astrocytes make the bulk of it.
What is really interesting is that they highlighted some of the activated microglial signatures that occur during tau pathology, and these were decreased in the astrocyte ApoE4-deleted mice. Also interesting, but still requiring further study, is that many of these effects were strongest or only present in female mice. A good explanation for this is not apparent from the current data.
Overall, this adds to the previous landmark study that the Holtzman lab published on global effects of ApoE in tau models, and suggests that astrocytes are major mediators of this, though conditional deletion of ApoE in microglia is being pursued by several labs and it effects remain to be seen.
National Institutes of Health
This paper by Wang et al. describes multiple effects of inducibly removing astrocytic APOE isoforms (APOE3 or APOE4) in a tau P301S mouse model. This model system is powerful in that it allows the researchers to isolate the contribution of astrocytic APOE isoforms to pathology and molecular signatures of neurodegeneration.
Wang et al. discover that inducible knockdown of APOE4, and not APOE3, results in a number of protective effects in a tauopathy mouse model. These many effects include molecular phenotypes such as less astrocytic activation, altered oligodendrocyte transcription, changes in excitatory neuron populations, and decreased phagocytosis of synapses by microglia.
This study re-emphasizes and expands on previous work by the Holtzman lab and others to make very clear that astrocytic APOE4 represents a toxic gain of function. This gain of toxic function can impact cell physiology in both cell autonomous and cell non-autonomous ways. It will be interesting to see if the same toxic gain-of-function effect holds true for APOE4’s influence on amyloid-centric, or other disease-centric models.
This paper presents a number of intriguing hypotheses about interactions between cell types that are impacted by genotype and presence of APOE. These are ripe for future mechanistic investigations. Given APOE’s role in lipid metabolism, and the involvement of lipids in nearly every cellular process, it is possible that lipid state is a key mediator of astrocytic APOE’s inter- and intracellular effects.
One interesting theme throughout the data is that knockdown of APOE4 has a stronger effect on female animals than males. This echoes the literature in both mouse models and humans—in the context of tauopathies, APOE4’s detrimental effects are far greater on females than males (Nov 2019 news). It will be interesting to see if this correlation remains true for diseases other than tauopathies for which APOE4 increases risk.
The strong protective effect of astrocytic APOE knockdown identified in this work supports the exploration of therapeutics aimed at reducing APOE levels. A challenge does arise in designing cell type-specific therapeutics. More investigation into the cell-type-specific effects of APOE knockdown will be essential in developing targeted and nuanced therapeutics or preventative approaches.
Genentech
This new study fits with previous work from the Holtzman lab showing that ApoE4 knock-in exacerbates the neuroinflammation and neurodegeneration that occurs in this tauopathy model, while ApoE4 reduction by antisense oligonucleotide treatment opposes these effects (Shi et al., 2017; Litvinchuk et al., 2021).
One open question was whether ApoE4 derived from specific cell types was of particular importance to driving neuroinflammation and degeneration. In particular, while astrocytes are a major source of ApoE, activated microglia as found in the tauopathy model exhibit a striking induction of ApoE expression, raising the hypothesis that microglia-derived ApoE could be playing a key role during pathology.
The new findings that the removal of ApoE4 selectively in astrocytes is sufficient to reduce neurodegeneration and associated glial activation argue against a key role for microglia ApoE during pathology in this model. Therefore, the role of activated microglia in mediating tau-dependent pathology (Shi et al., 2019; Mancuso et al., 2019) is likely not so much via to production of ApoE, but rather as a mediator of neuronal damage in response to the harmful inflammatory state of the brain that is caused by tau pathology, and exacerbated by ApoE4.
A remaining goal for the field is to gain a better mechanistic understanding of the actions of ApoE4 on the interactions between tau pathology, glial activation, and synapse and neuronal loss.
References:
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, Tsai RM, Spina S, Grinberg LT, Rojas JC, Gallardo G, Wang K, Roh J, Robinson G, Finn MB, Jiang H, Sullivan PM, Baufeld C, Wood MW, Sutphen C, McCue L, Xiong C, Del-Aguila JL, Morris JC, Cruchaga C, Alzheimer’s Disease Neuroimaging Initiative, Fagan AM, Miller BL, Boxer AL, Seeley WW, Butovsky O, Barres BA, Paul SM, Holtzman DM. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017 Sep 28;549(7673):523-527. Epub 2017 Sep 20 PubMed.
Litvinchuk A, Huynh TV, Shi Y, Jackson RJ, Finn MB, Manis M, Francis CM, Tran AC, Sullivan PM, Ulrich JD, Hyman BT, Cole T, Holtzman DM. Apolipoprotein E4 Reduction with Antisense Oligonucleotides Decreases Neurodegeneration in a Tauopathy Model. Ann Neurol. 2021 May;89(5):952-966. Epub 2021 Feb 24 PubMed.
Shi Y, Manis M, Long J, Wang K, Sullivan PM, Remolina Serrano J, Hoyle R, Holtzman DM. Microglia drive APOE-dependent neurodegeneration in a tauopathy mouse model. J Exp Med. 2019 Nov 4;216(11):2546-2561. Epub 2019 Oct 10 PubMed.
Mancuso R, Fryatt G, Cleal M, Obst J, Pipi E, Monzón-Sandoval J, Ribe E, Winchester L, Webber C, Nevado A, Jacobs T, Austin N, Theunis C, Grauwen K, Daniela Ruiz E, Mudher A, Vicente-Rodriguez M, Parker CA, Simmons C, Cash D, Richardson J, NIMA Consortium, Jones DN, Lovestone S, Gómez-Nicola D, Perry VH. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain. 2019 Oct 1;142(10):3243-3264. PubMed.
Icahn School of Medicine at Mount Sinai
Icahn School of Medicine at Mount Sinai
The new study in Neuron by Wang and colleagues, led by David Holtzman, Washington University, St. Louis, strengthens the evidence that APOE4 acts not only by gain of toxic function, but that APOE4 promotes multiple features of the classical Alzheimer’s disease (AD) phenotype. The title of the paper, “Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia,” communicates succinctly the important conclusions for which compelling mouse model data are presented.
Wang et al. describe use of the MAPT P301S tauopathy mouse line as the backbone for generation of an inducible cre-lox based system for APOE isotype expression and knockout (Tau/Aldh1l1-CreERT2/apoE3flox/flox or Tau/Aldh1l1-CreERT2/apoE4flox/flox). At 5.5 months of age, after the onset of tau pathology, they administered tamoxifen or vehicle and compared mice at 9.5 months of age. Removing astrocytic APOE4 markedly reduced tau-mediated neurodegeneration and decreased phospho-tau pathology. Single-nucleus RNA sequencing analysis revealed striking gene expression changes in all cell types, with astrocytic APOE4 removal decreasing disease-associated gene signatures in neurons, oligodendrocytes, astrocytes, and microglia. An exciting additional observation was that removal of astrocytic APOE4 decreased tau-induced synaptic loss and microglial phagocytosis of synaptic elements, suggesting that astrocytic ApoE plays a key role in synaptic degeneration. There is ample documentation that synaptic integrity—not severity of proteinopathy—is the best correlate of cognition during either aging or AD.
There are also already strong independent data indicating that APOE4 apparently promotes parenchymal and cerebrovascular amyloid accumulation directly, while promoting tauopathy both via acceleration of amyloidosis as well as by direct actions of ApoE4 on tauopathy. ApoE4 disrupts the blood-brain barrier in analogous dual mechanisms, i.e., apparently both by impairing pericyte viability and by promoting cerebrovascular amyloidosis that promotes degeneration of the medial layer of the vessel wall.
Though the new Wang et al. story involves astrocyte-derived ApoE4, we and others have observed roles played by microglial APOE in the phenotypic switching from homeostatic to disease-associated microglia (DAM). We have recently observed that APOE-related induction of the DAM phenotype can be caused by molecular pathways via TYROBP even in the absence of TREM2 (Audrain et al., 2020). This is in addition to the role, well-documented by several groups, indicating that TREM2 induces DAM via ApoE.
Koldamova and colleagues have reported that APOE4 acts more potently in microglial phenotype switching than does APOE3 (Fitz et al., 2020). Wang et al. confirm that tauopathy can drive the switch from homeostatic microglia toward DAM, but they report the unexpected observation that knocking down astrocytic APOE4 can attenuate the microglial DAM phenotype. RNA sequencing data have implicated APOE transcripts in microglia, but, as far as we are aware, Wang et al. present the first evidence that the isotype of the ApoE protein secreted from astrocytes can modulate phenotypic switching in microglia. It is tempting to speculate that this scenario is underpinned by paracrine and autocrine signaling pathways wherein microglial cell-surface molecules are triggered by ApoE (and/or other ligands), thereby stimulating intramicroglia transcription of DAM genes.
The potential clinical impacts of this new data are several. First, depletion of astrocytic APOE4 acts to protect neurons and synapses based on structural data. Electrophysiological characterization of the Wang et al. mice depleted of astrocytic APOE4 would be an interesting follow-up.
Moreover, this is now a third molecule that modulates synaptophagy by microglia. Hong et al. (2016) and Shi et al. (2017) identified the Aβ soluble oligomers and CR3 as two key players in this process, one that all evidence suggests is relevant for cognitive reserve and/or functional resilience, despite the otherwise clinically important severities of the proteinopathies. Given that depletion of the most neurotoxic soluble Aβ oligomer strains may be important for clinically meaningful benefit in response to passive immunotherapy with, e.g., aducanumab, BAN2401, and/or donanemab (reviewed in Gandy and Ehrlich, 2021, J Exp Med, in press), the discovery that astrocytic APOE4 mitigates synaptophagy may be especially relevant to AD, since half or more of late-onset sporadic AD is associated with the presence of at least one APOE4 allele. Identification of APOE4 carriers as those most likely to show clinically meaningful benefit following preclinical initiation of Aβ-reducing interventions would move us closer to the identification of those in whom the risk and expense of Aβ-reducing passive immunotherapy would be potentially justifiable.
One key piece that we are still missing is an understanding of how to intervene when genetic risk for AD is linked to those subjects in whom progression of cognitive decline is apparently underpinned by a genetic-based remission of amyloidosis over time, rather than the more typical progressive clinical decline that goes hand in hand with accumulating amyloidosis. The classical clinicopathological correlations among progressive amyloidosis, progressive tauopathy, and cognitive decline are now included as an essential troika of entry criteria for some Aβ-reducing passive immunotherapies. The outliers whose cognitive decline does not conform to this classical triad are ineligible for Aβ-reducing trials. However, Wang et al.’s Neuron data raise the possibility that the APOE4 carriers among those with noncanonical CR3-linked cognitive decline associated with “vanishing amyloidosis” might respond to depletion of astrocytic APOE4 (Thambisetty et al., 2012; Gandy et al., 2013).
When contemplating how APOE might be therapeutically reduced, Holtzman has championed the use of passive immunotherapy to target ApoE rather than Aβ (Xiong et al., 2021). The new data on the benefit of APOE4 depletion should add momentum to anti-ApoE passive immunotherapies. Yet another way of targeting APOE is already in human clinical trials: Crystal and colleagues at Weill Cornell Medical College have reported rodent and primate data suggesting that infusion and seeding of AAV-APOE2 into the subarachnoid space of APOE4 carriers might neutralize the untoward consequences of APOE4 for neurons, synapses, and pericytes (Zhao et al., 2016; Rosenberg et al., 2018). No formal reports from this Phase 1 trial have yet emerged, and recruitment of symptomatic APOE4 homozygotes continues. At the very least, linking common and readily identifiable genetic risks (such as APOE genotype) to clinically relevant molecular pathogenesis and patient selection might well move us closer to the eventual goal of prevention or arrest of progressive cognitive decline associated with AD.
References:
Audrain M, Haure-Mirande JV, Mleczko J, Wang M, Griffin JK, St George-Hyslop PH, Fraser P, Zhang B, Gandy S, Ehrlich ME. Reactive or transgenic increase in microglial TYROBP reveals a TREM2-independent TYROBP-APOE link in wild-type and Alzheimer's-related mice. Alzheimers Dement. 2021 Feb;17(2):149-163. Epub 2020 Dec 12 PubMed.
Fitz NF, Wolfe CM, Playso BE, Biedrzycki RJ, Lu Y, Nam KN, Lefterov I, Koldamova R. Trem2 deficiency differentially affects phenotype and transcriptome of human APOE3 and APOE4 mice. Mol Neurodegener. 2020 Jul 23;15(1):41. PubMed.
Gandy S, Haroutunian V, Dekosky ST, Sano M, Schadt EE. CR1 and the "vanishing amyloid" hypothesis of Alzheimer's disease. Biol Psychiatry. 2013 Mar 1;73(5):393-5. PubMed.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016 May 6;352(6286):712-6. Epub 2016 Mar 31 PubMed.
Rosenberg JB, Kaplitt MG, De BP, Chen A, Flagiello T, Salami C, Pey E, Zhao L, Ricart Arbona RJ, Monette S, Dyke JP, Ballon DJ, Kaminsky SM, Sondhi D, Petsko GA, Paul SM, Crystal RG. AAVrh.10-Mediated APOE2 Central Nervous System Gene Therapy for APOE4-Associated Alzheimer's Disease. Hum Gene Ther Clin Dev. 2018 Mar;29(1):24-47. Epub 2018 Mar 13 PubMed.
Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. 2017 May 31;9(392) PubMed.
Thambisetty M, An Y, Nalls M, Sojkova J, Swaminathan S, Zhou Y, Singleton AB, Wong DF, Ferrucci L, Saykin AJ, Resnick SM, . Effect of Complement CR1 on Brain Amyloid Burden During Aging and Its Modification byAPOEGenotype. Biol Psychiatry. 2012 Sep 27; PubMed.
Xiong M, Jiang H, Serrano JR, Gonzales ER, Wang C, Gratuze M, Hoyle R, Bien-Ly N, Silverman AP, Sullivan PM, Watts RJ, Ulrich JD, Zipfel GJ, Holtzman DM. APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci Transl Med. 2021 Feb 17;13(581) PubMed.
Zhao L, Gottesdiener AJ, Parmar M, Li M, Kaminsky SM, Chiuchiolo MJ, Sondhi D, Sullivan PM, Holtzman DM, Crystal RG, Paul SM. Intracerebral adeno-associated virus gene delivery of apolipoprotein E2 markedly reduces brain amyloid pathology in Alzheimer's disease mouse models. Neurobiol Aging. 2016 Aug;44:159-72. Epub 2016 Apr 30 PubMed.
Universitat Autònoma de Barcelona
Two comments:
1) This study, however impressive, has a limitation: There is no Aβ pathology. Aβ uptake by astrocytes damages their endolysosomal system (Sanchez-Mico et al., 2021). Thus, the question is: How would astrocytes damaged by Aβ respond to Tau pathology in the context of different APOE alleles? It is essential to consider the interplay between Tau and Aβ in order to establish the contribution of astrocytes to Alzheimer's disease.
2) As recommended in a recent consensus article about reactive astrocytes (Escartin et al., 2021), the terminology "homeostatic" and "senescent" or even "reactive" should be avoided for any transcriptional cluster until functions are clarified. Plausibly, the so-called "reactive" and "senescent" clusters encompass a mixed set of adaptive and maladaptive responses that respectively maintain and disrupt astrocytic homeostasis.
References:
Sanchez-Mico MV, Jimenez S, Gomez-Arboledas A, Muñoz-Castro C, Romero-Molina C, Navarro V, Sanchez-Mejias E, Nuñez-Diaz C, Sanchez-Varo R, Galea E, Davila JC, Vizuete M, Gutierrez A, Vitorica J. Amyloid-β impairs the phagocytosis of dystrophic synapses by astrocytes in Alzheimer's disease. Glia. 2021 Apr;69(4):997-1011. Epub 2020 Dec 7 PubMed.
Escartin C, Galea E, Lakatos A, O'Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhäuser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen WT, Cohen-Salmon M, Cunningham C, Deneen B, De Strooper B, Díaz-Castro B, Farina C, Freeman M, Gallo V, Goldman JE, Goldman SA, Götz M, Gutiérrez A, Haydon PG, Heiland DH, Hol EM, Holt MG, Iino M, Kastanenka KV, Kettenmann H, Khakh BS, Koizumi S, Lee CJ, Liddelow SA, MacVicar BA, Magistretti P, Messing A, Mishra A, Molofsky AV, Murai KK, Norris CM, Okada S, Oliet SH, Oliveira JF, Panatier A, Parpura V, Pekna M, Pekny M, Pellerin L, Perea G, Pérez-Nievas BG, Pfrieger FW, Poskanzer KE, Quintana FJ, Ransohoff RM, Riquelme-Perez M, Robel S, Rose CR, Rothstein JD, Rouach N, Rowitch DH, Semyanov A, Sirko S, Sontheimer H, Swanson RA, Vitorica J, Wanner IB, Wood LB, Wu J, Zheng B, Zimmer ER, Zorec R, Sofroniew MV, Verkhratsky A. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021 Mar;24(3):312-325. Epub 2021 Feb 15 PubMed.
NYU Langone
NYU
This is an exciting set of data from the Holtzman group that adds another piece to the puzzle of APOE’s role as a risk factor for Alzheimer’s Disease (AD). In this paper, Wang et al. use the P301S mouse model of AD, expressing either ApoE3 or ApoE4, and genetically remove the APOE isoforms using a Cre/flox system with the pan-astrocytic, astrocyte-specific Aldh1l1 promoter as the Cre driver.
They find that removing APOE4 specifically in astrocytes reduces brain atrophy, slows accumulation of phosphorylated tau, and improves behavioral deficits. Using single-nucleus RNA-sequencing, they find that APOE4 expression not only has drastic consequences for astrocytes, but also affects gene expression in non-astrocytic cells. Indeed, they find a shift in the cluster composition of neurons, including a surprising APOE4-dependent increase in Rorb-positive neurons, and among microglia they find an increase in major histocompatibility complex (MHC)-expressing cells, which are in turn repressed by removing APOE from astrocytes, which themselves show a shift in their relative subtype abundance. Indeed, ApoE4-carrying mice show an increase in numbers of astrocytes expressing genes classically defined as “reactive,” including Gfap and Vim, which are repressed by the genetic removal of APOE4 from astrocytes. Given the abundance of studies now highlighting the single cell heterogeneity of astrocyte (and microglia) responses to both acute insults and chronic neurodegenerative diseases, the authors also investigated these discrete astrocyte responses across different APOEe isoform-expressing mouse models.
The use of single cell/nucleus RNA-sequencing is a powerful approach to study acute and chronic diseases in a cell-type-wide and unbiased manner. Particularly the bioinformatic integration of samples across different conditions, as applied here, allows scientists to pinpoint the disease progression of once-healthy cells. This paper and others have shown how astrocytes can be quite heterogenous and how an insult can add an additional layer of complexity. While the numbers of cells collected is quite low, they add to the growing repertoire of astrocyte single-cell datasets that can be used for ongoing meta-analysis and integration to further understand the subtle and heterogeneous transcriptomic changes that may depend on time, sex, or disease-associated mutations.
Indeed, adding additional time points, pseudotime, and pseudobulk analyses, as well as increasing the number of sequenced astrocytes will help us understand the reactive transition astrocytes can undergo. Similarly, adding additional modalities, such as spatial transcriptomics, and performing multimodal data integration are powerful tools to unravel how both the subtype and anatomical location of an astrocyte determines its response to an inflammatory insult.
All in all, this is another beautiful example of how a cell-type-specific intervention can have non-cell autonomous consequences, highlighting the need to consider the multicell type composition of the brain when studying disease. The use and integration of multimodal sequencing tools, such as single cell/nucleus RNA-sequencing and spatial transcriptomics, will help us uncover what and where things go awry in brain cells in disease, and aid us in identifying novel therapeutic strategies.
Make a Comment
To make a comment you must login or register.