ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
Quick Links
A flurry of recent studies implicating microglia as key players in neurodegenerative diseases, particularly Alzheimer’s, has cued many labs to try to figure out how these cells function in health, and how they change in disease. Adding a piece to the puzzle, scientists led by Oleg Butovsky of Brigham and Women’s Hospital in Boston report in the September 19 issue of Immunity that two genes with variants known to alter Alzheimer’s risk—APOE and TREM2—orchestrate the cells’ passage from a homeostatic state to a disease-associated state, at least in mice.
- In mouse models, microglia adopt a neurodegeneration-like transcription profile.
- Degenerating neurons trigger, and APOE and TREM2 mediate, this transition.
- Blocking APOE in microglia prevents the switch.
“This is a fascinating paper. It provides substantial evidence for an APOE-TREM2 pathway in mouse microglia that senses apoptotic neurons and induces a ‘neurodegenerative’ phenotypic switch,” wrote Christopher Glass, University of California, San Diego, in an email to Alzforum.
Microglia continuously survey the brain to preserve order and stability, providing trophic support for neurons, phagocytosing cellular debris, and responding to foreign invaders. Absent injury or disease, microglia express a batch of genes controlled by transforming growth factor β (TGFβ) signaling profile of microglia (Butovsky et al., 2014; Gosselin et al., 2014). Alas, when disease, stress, or injury strike, microglia take on other identities, marked by expression of different sets of genes.
Researchers have begun to characterize microglial transcriptome changes in neurodegeneration (Jun 2017 news; Crotti and Ransohoff, 2016, for review). Ido Amit and colleagues at the Weizmann Institute, Rehovot, Israel, for example, recently described a subgroup of disease-associated microglia, aka DAMs, in the brains of 5xFAD mice (Jun 2017 news). DAMs express low levels of homeostatic, but high levels of lipid metabolism, phagocytosis, and AD-relevant genes, including APOE and TREM2.

Altered State: In healthy mouse brains, microglia assume a homeostatic state in which TGFβ controls gene expression (blue). When triggered by degenerating neurons, TREM2 and APOE signaling transform gene expression (red). Whether this switch helps or harms brain function likely depends on the type and stage of disease. [Courtesy of Immunity, Krasemann et al., 2017.]
Hoping to unravel what’s behind this type of transformation, co-first authors Susanne Krasemann and Charlotte Madore surveyed the expression patterns of microglia derived from mouse models of AD (APP-PS1), multiple sclerosis (experimental autoimmune encephalomyelitis [EAE]), and amyotrophic lateral sclerosis (SOD1-G93A) over time. “We asked the big question: How do these cells transform; how is this regulated?” said Butovsky. The researchers used the microglial-specific antibody anti-FCRLS to pull microglia from brains and spinal cords dissected at different stages of disease. They then analyzed the cells’ RNAs using a custom-made chip covering 400 genes uniquely or highly expressed in microglia, plus 40 inflammation and phagocytic genes known to be affected in the mice. A clustering algorithm sorting the results in an unbiased way highlighted two groups of genes: those whose expression dropped with disease stage and age, including 68 homeostatic genes, and those whose expression rose, including 28 inflammatory genes (image below). The researchers named the state associated with this profile “microglial neurodegenerative phenotype,” or MGnD. In the EAE mice, the MGnD profile surfaced in the acute phase of disease, and the homeostatic profile returned during the recovery period.
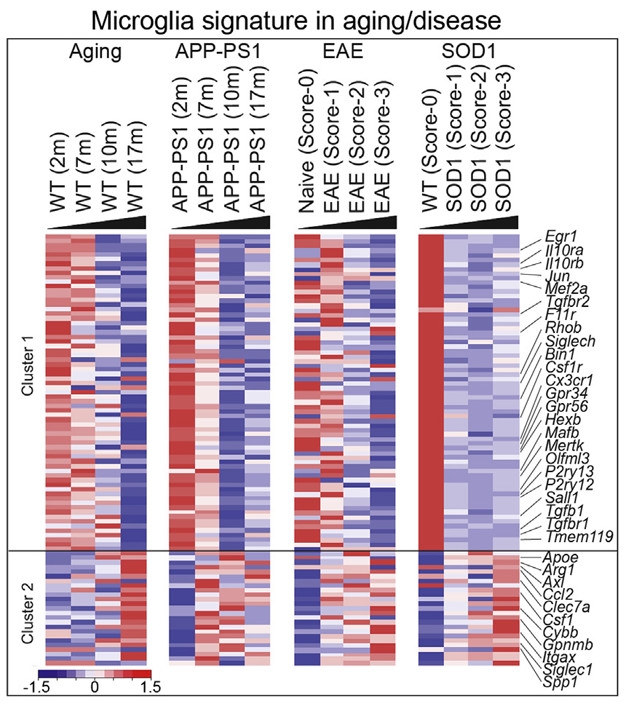
Transformation Within: As disease, or aging, progresses, the gene expression profile of microglia switches to a neurodegenerative phenotype (MGnD). Cluster 1, which includes homeostatic genes, fades; cluster 2, e.g. APOE, rises. [Courtesy of Immunity, Krasemann et al., 2017.]
Using pathway analyses to relate the expression changes to each other, the researchers homed in on APOE induction and TGFβ suppression as the major upstream regulators of MGnD. The TGFβ-dependent genes whose expression waned included Olfml3, P2ry12, Tmem119, Mef2a, Jun, Sall1, while APOE, Axl, and Clec7a came up most strongly.
Importantly, the authors distinguished the MGnD state from classic microglial activation. While treating mice with microglial activators, such as lipopolysaccharide or interferon-γ, suppressed homeostatic microglial genes, these activated profiles lacked APOE induction and differed in other ways from MGnD, as well. “It’s a striking difference. LPS is not recapitulating disease,” said Butovsky.
How do MGnD microglia compare to the DAM described by Amit and colleagues? “I think they are very similar. Whether they are absolutely identical one cannot say because they used different models and different conditions,” said Amit’s co-author Michal Schwartz, also at the Weizmann Institute. Alison Goate of Mount Sinai School of Medicine in New York agreed.
Where do those MGnD microglia appear in the brain? The authors stained brain sections of 24-month-old APP-PS1 mice with P2ry12 antibodies as a marker of homeostatic microglia, and Clec7a as a marker of MGnD. P2ry12-/Clec7a+ cells encircled plaques with damaged neurites themselves, while P2ry12+/Clec7a- cells blanketed the surrounding tissue a bit farther away (image below).

Drawn to Death.
MGnD microglia (red) crowd a plaque with damaged neurites (stained on the right) in APP-PS1 mouse brain tissue, while homeostatic microglia (green) remain at the edges. [Immunity, Krasemann et al., 2017.]
Isolating Clec7a+ and P2ry12+ microglia to analyze their transcription pattern confirmed that Clec7a+ cells had a MGnD and P2ry12+ cells a homeostatic phenotype. In human AD brain tissue, too, the P2ry12 signal was dim in neuritic plaques, but plaques devoid of injured neurites lit up with this marker. This suggested to the authors that microglia retain their homeostatic identity in the absence of neuronal damage.
“We asked: What is the common feature associated with MGnD? Is it dying neurons?” said Butovsky. To check this out, the authors injected fluorescently labeled, apoptotic neurons into the cortices and hippocampi of wild-type mice. Microglia engulfed the dying cells, and those that did, boosted ApoE expression as early as three hours post-injection. In contrast, injection of E. coli or inert beads failed to induce ApoE. By separating phagocytosing from non-phagocytosing microglia in mice injected with dying neurons, the researchers determined that only microglia loaded with debris expressed the MGnD signature. Consistent with this, they found the MGnD fingerprint in microglial transcriptome data from mouse models of several neurodegenerative disorders, but not in models of other neurological conditions that lack neuronal death, such as pain. Goate was intrigued by the finding and speculated on the nature of the trigger. “It could be membrane damage, such as injury to cell membranes caused by Aβ toxicity, aging, or synaptic pruning,” she speculated.
To probe the underlying signaling, the researchers began manipulating genes. Suspecting APOE was involved, they examined microglial expression profiles in APOE knockout mice. When “fed” dying neurons, these microglia phagocytosed them but did not switch to the MGnD phenotype. This appeared to be cell-autonomous, because when the researchers suppressed APOE only in microglia, they saw the same inability to transition to MGnD. To see how this failure to switch to MGnD might affect axonal injury situations, the authors cut a facial nerve in APOE-deficient mice and examined the damaged cells seven days later. The neurons of mice lacking ApoE, either everywhere or only in microglia, fared better than those of wild-type. Could ApoE on its own flip the MGnD switch? The authors injected recombinant ApoE into the hippocampi of ApoE knockout mice and, voila, homeostatic gene expression dropped and MGnD expression climbed in the local microglia.
Next, the scientists eyed TREM2 as a possible accomplice because TREM2 binds ApoE and senses the kinds of lipids damaged cells spill. Indeed, knocking out TREM2 suppressed the MGnD profile, locking microglia in APP-PS1 and SOD1 mice in homeostasis. Interestingly, in the APP-PS1 mice, plaque load dropped at four months of age. As with ApoE deficiency, TREM2 deficiency protected neurons whose axons had been cut. Consistent with results in 5XFAD mice, in which TREM2-deficient microglia don’t descend on Aβ plaques (Feb 2015 news), the researchers saw fewer microglia crowding around plaques in AD postmortem brains of carriers of the R47H or R62H TREM2 loss-of-function mutations than in patients without those mutations. The carriers’ tissue also stained more brightly with antibodies against the homeostatic protein Tmem119.
While Amit and colleagues had also implicated TREM2 in DAM activation, they had only seen it later in the transformation process, after homeostatic genes were suppressed. Schwartz nevertheless thinks that “for APOE to be fully induced you absolutely need TREM2.” Unfortunately, a robust causal link between the two has yet to be established. “I wish Krasemann and colleagues had done experiments with added ApoE in TREM2 knockouts, too. That could suggest if TREM2 is upstream or not,” said Goate.
Another question is whether ApoE acts by binding TREM2 on the surface of microglia or by acting as an intracellular signaling molecule. “I think there are probably two independent programs,” said Goate. “It’s a big question—we don’t know much about ApoE upregulation in microglia.” Much of the work on ApoE to date has focused on astrocytes or Aβ metabolism, she added. Goate said she is particularly intrigued by what the effects of different APOE risk variants might be on microglia.
Does the MGnD phenotype fight or fuel neurodegeneration? Genetics suggests TREM2 is protective, Anna Pimenova, Edoardo Marcora, and Goate write in an accompanying commentary. Partial loss-of-function mutations boost AD risk (Nov 2012 news). TREM2 deficiency reportedly keeps microglia stuck in a homeostatic state, which prevents microglia from swooping in on plaques, phagocytosing amyloid and damaged cells, and proliferating (Jul 2016 news; May 2017 news). Also, a protective allele in the gene SPI1 is associated with a MGnD-like expression signature (Jun 2017 news). “This would argue MGnD microglia are protective, at least early on,” said Goate. Schwartz agreed. “I think the transition is needed for coping with disease,” she said, “Maybe the problem is that the transition occurs too late.”
Yet Butovsky’s new study suggests MGnD can be detrimental. The authors cite other findings consistent with this. For example, the CD33 AD risk allele is linked to high TREM2 expression (Oct 2015 news). “MGnD microglia may not always be bad,” Butovsky acknowledged, adding, “They could be beneficial at later stages of AD.” He suspects late, rather than early, benefits because of the low plaque load his team observed in young APP-PS1 mice lacking TREM2 (Jay et al., 2015; Jay et al., 2017). More work is needed to sort out this question.
Do MGnD microglia play similar roles in different neurodegenerative conditions? “It is puzzling that the microglial genetic risk is seen strongly only in AD. Parkinson’s disease and ALS have a microglial component, but it’s less strong. Maybe there is more widespread death throughout the brain in AD,” speculated Goate. She also noted that the MGnD phenotype may affect risk in some diseases, while changing the rate of progression in others. This would jibe with a recent role of ApoE in exacerbating microglial responses and accelerating progression in the presence of tau pathology (Sep 2017 news).
Butovsky believes the MGnD switch likely affects different diseases differently. For example, the switch to MGnD in EAE and SOD1 mice eliminated microglias’ ability to rein in T cell proliferation. This is a crucial function in multiple sclerosis, where rogue T cells damage myelin.
Looking ahead, UCSD’s Glass would like to see confirmation of the MGnD phenotype in human microglia. For his part, Butovsky is excited about the possibilities of targeting ApoE. “Can we manipulate ApoE focally, perhaps specifically in microglia?” he asked.—Marina Chicurel
References
News Citations
- What Makes a Microglia? Tales from the Transcriptome
- Hot DAM: Specific Microglia Engulf Plaques
- TREM2 Buoys Microglial Disaster Relief Efforts in AD and Stroke
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
- TREM2 Helps Phagocytes Gobble Up Aβ Coated in Antibodies
- Paper Alert: TREM2 Crucial for Microglial Activation
- Microglial Master Regulator Tunes AD Risk Gene Expression, Age of Onset
- Alzheimer’s Risk Genes Interact in Immune Cells
- ApoE4 Makes All Things Tau Worse, From Beginning to End
Research Models Citations
Paper Citations
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014 Jan;17(1):131-43. Epub 2013 Dec 8 PubMed.
- Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, Glass CK. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014 Dec 4;159(6):1327-40. PubMed.
- Crotti A, Ransohoff RM. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity. 2016 Mar 15;44(3):505-15. PubMed.
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM, Lamb BT. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med. 2015 Mar 9;212(3):287-95. Epub 2015 Mar 2 PubMed.
- Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, Lamb BT, Landreth GE. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer's Disease. J Neurosci. 2017 Jan 18;37(3):637-647. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O'Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017 Sep 19;47(3):566-581.e9. PubMed.
- Pimenova AA, Marcora E, Goate AM. A Tale of Two Genes: Microglial Apoe and Trem2. Immunity. 2017 Sep 19;47(3):398-400. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of California San Diego
This is a fascinating paper. It provides substantial evidence for an ApoE-Trem2 pathway in mouse microglia that senses apoptotic neurons and induces a "neurodegenerative" phenotypic switch.
A particularly interesting finding is the evidence for a cell-autonomous role of microglia ApoE in driving this phenotype, based on its microglia-specific deletion.
A challenging aspect of this work is to clearly relate it to genetic studies in humans that indicate pathogenic consequences of loss of Trem2 function and protective consequences of lowered PU.1 expression. The loss of the homeostatic signature of microglia following phagocytosis of injected apoptotic neurons is intriguing but hard to interpret at this stage, in part because macrophage phagocytosis of apoptotic cells in the periphery is usually with tissue homeostatic roles.
It is thus not clear at this stage whether the "neurodegenerative" phenotype is pathogenic or an adaptive response. The main findings will thus certainly provoke substantial discussion and follow-up experiments about roles of this pathway in neurodegenerative disease. It will be particularly important to investigate whether this pathway operates in a similar manner in human microglia.
Make a Comment
To make a comment you must login or register.