A Putrid Problem: Astrocytic Urea Cycle in Alzheimer’s?
Quick Links
What determines whether astrocytes play a beneficial or detrimental role in Alzheimer’s disease? It’s surely complicated, but a study in the June 10 online Cell Metabolism lays some of the blame on the cells’ urea metabolism, a biochemical pathway that detoxifies ammonia. Researchers led by Justin Lee at the Institute for Basic Science, Daejeon, and Hoon Ryu at the Korea Institute of Science and Technology, Seoul, both in the Republic of Korea, found that the breakdown of Aβ in astrocytes produces ammonia, fueling the urea cycle and creating toxic side products, in particular the stinky organic compound putrescine. Excess putrescine is converted into the inhibitory neurotransmitter GABA, dampening synaptic transmission and impairing memory. In a mouse model of amyloidosis, blocking putrescine production brought GABA levels down to normal and rescued learning and memory.
- When exposed to Aβ, astrocytes fire up the urea cycle, producing excess putrescine.
- Putrescine is converted into GABA, which inhibits synaptic transmission and blunts memory in amyloidosis mice.
- Inhibiting the enzyme that makes putrescine rescues mouse synapses and memory.
Sagar Gaikwad at the University of Texas Medical Branch in Galveston called the study important. “A deeper understanding of urea metabolism in reactive astrocytes may prove to be critical in understanding mechanisms of neurodegeneration,” he wrote (full comment below).
Previously, Lee and colleagues had reported that reactive astrocytes in amyloidosis mice pump out GABA. Suppressing GABA production rescued synaptic excitability and memory. In AD brain, as well, this group found astrocytic GABA to be elevated (Nov 2012 conference news; Jul 2014 news). Other work has recorded an excess of polyamines such as arginine and putrescine in AD brain (May 2021 news; May 2021 news; Polis et al., 2021).
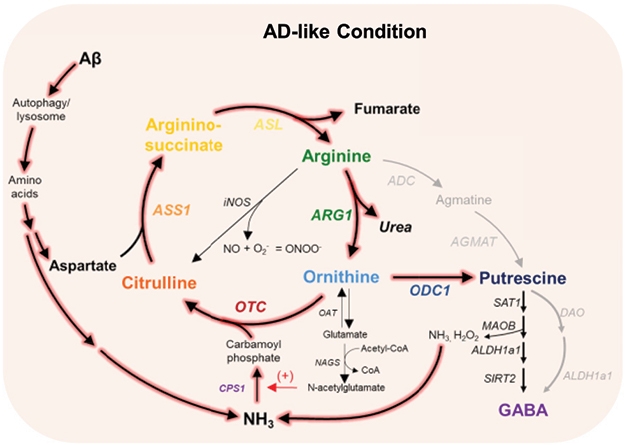
From Aβ to GABA. In mouse astrocytes, autophagy breaks down Aβ into aspartate and ammonia (NH3); these feed into the urea cycle and produce excess putrescine, which in turn becomes GABA. [Courtesy of Ju et al., Cell Metabolism.]
To figure out what was behind this excess GABA, first author Yeon Ha Ju at IBS isolated primary astrocytes from the cortices of neonatal wild-type mice. Because putrescine’s biochemical precursors, arginine and ornithine, are part of the urea cycle, Ju and colleagues examined how this cycle changed in response to Aβ. In astrocytes exposed to 1 μM synthetic oligomeric Aβ42 for five days, the urea cycle revved up twofold. In particular, the enzyme ornithine decarboxylase 1 (ODC1) was elevated, converting ornithine to putrescine rather than to its normal end product, glutamate.
How did Aβ boost the urea cycle? Aβ-treated cells accumulated aspartate and ammonia, both of which feed into the cycle. Inhibiting autophagy dropped the amount of these metabolites back to nearly control levels, suggesting that autophagic degradation of Aβ produces the toxic waste products driving the cycle.
Suppressing urea cycle enzymes, particularly ODC1, using short hairpin RNAs halved the amount of putrescine and brought GABA back to levels seen in control astrocytes. In addition, inhibiting ODC1 squelched hydrogen peroxide (H2O2), a toxic byproduct of GABA production that contributes to oxidative stress. It also lowered ammonia, another byproduct of GABA production. That suggested the existence of a toxic feedback loop in response to Aβ, where synthesis of putrescine and GABA produces ammonia that further drives the cycle.

Aβ Boosts Putrescine. In wild-type mouse hippocampus (left), there is almost no ornithine decarboxylase 1 (green), but in reactive astrocytes of APP/PS1 mice (right), the enzyme that makes putrescine is highly expressed. [Courtesy of Ju et al., Cell Metabolism.]
Would this happen in vivo? In wild-type mice, immunostaining revealed almost no ODC1, but in reactive astrocytes in the hippocampi of APP/PS1 mice, the enzyme was abundant. As in cultured astrocytes, inhibiting astrocytic ODC1 with shRNA brought putrescine and GABA back to normal levels. It also calmed astrocytes, as seen by less expression of the inflammatory marker GFAP and a return to a homeostatic cell shape with shorter processes. Synaptic transmission rebounded, and the mice learned as well as wild-type in the Y-maze, a measure of short-term spatial memory. On the passive-avoidance test, a measure of long-term spatial memory, treatment improved performance, but not back to wild-type levels, the scientists reported.
“Based on our findings, we propose that ODC1 inhibition turns toxic reactive astrocytes into Aβ-detoxifying astrocytes in AD,” the authors wrote.
Surprisingly, four weeks after ODC1 silencing, the mice had about half as many neuritic plaques as did untreated APP/PS1 mice. Experiments in cultured astrocytes showed that inhibiting ODC1 suppressed amyloidogenic cleavage of amyloid precursor protein and favored non-amyloidogenic cleavage. The mechanism is unclear.
But are these findings relevant to AD? Co-author Junghee Lee at Boston University stained reactive astrocytes in postmortem AD and control hippocampi for urea cycle enzymes. As in mice, ODC1 was highly elevated in AD, about 2.5 times higher than in control brain.
ODC1 could be a potential therapeutic target, the authors suggest. A pharmacological ODC1 inhibitor, difluoromethylornithine (DFMO), exists, and is being evaluated in certain types of cancer, but is quite toxic. The data suggest the need to develop better ODC1-inhibition strategies, such as small-molecule inhibitors or antisense oligos, the authors note.
M. Kerry O’Banion at the University of Rochester Medical Center, New York, said the study tells a compelling story, but cautioned that the situation in the Alzheimer’s brain is more complicated than in these model systems. For example, neuroinflammation is known to affect astrocyte metabolism and may change the picture. Gaikwad agreed that other factors such as pathological tau, oxidative stress, and aging may be influencing astrocytes in AD brain. He suggested testing whether the changes in astrocyte urea metabolism relate to senescence, which he previously found contributed to neurotoxicity in AD (Jul 2021 news).—Madolyn Bowman Rogers
References
News Citations
- SfN: Glial-Neuronal Signaling and AD Pathology
- Does Glial Neurotransmission Impair Memory Circuits in Alzheimer’s?
- Better Living Through Polyamines?
- Polyamines–What Role in Neurodegeneration?
- Astrocytes Are Just Dying to Spread Tau
Research Models Citations
Paper Citations
- Polis B, Karasik D, Samson AO. Alzheimer's disease as a chronic maladaptive polyamine stress response. Aging (Albany NY). 2021 Apr 3;13(7):10770-10795. PubMed.
Further Reading
Primary Papers
- Ju YH, Bhalla M, Hyeon SJ, Oh JE, Yoo S, Chae U, Kwon J, Koh W, Lim J, Park YM, Lee J, Cho IJ, Lee H, Ryu H, Lee CJ. Astrocytic urea cycle detoxifies Aβ-derived ammonia while impairing memory in Alzheimer's disease. Cell Metab. 2022 Aug 2;34(8):1104-1120.e8. Epub 2022 Jun 22 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Texas Medical Branch
In this important study, Ju et al. showed that under normal conditions, astrocytic urea metabolism is non-cyclic, and that it becomes cyclic in reactive astrocytes upon exposure to cellular stress, i.e., Aβ.
It is well known that upon exposure to pathological stimuli, astrocytes become reactive and eliminate protein aggregates. However, the chronic cellular stress by Aβ and pathological Tau exposure leads to overactivation and the cells become neurotoxic reactive astrocytes. The current study is highly relevant because reactive astrocytes are widely found in many human neurodegenerative diseases, including AD (Liddelow et al., 2017).
In this context, we and others demonstrated that astrocyte senescence is linked to loss of beneficial function and gain of neurotoxic function that contributes to Alzheimer’s disease and frontotemporal dementia (Gaikwad et al., 2021; Cohen and Torres, 2019). It would be interesting to understand whether cyclic urea metabolism is linked to astrosenescence and AD brain pathology.
Additionally, the authors demonstrated that astrocytic ornithine decarboxylase-1 (ODC1) is an important upstream enzyme for excessive production of GABA—the byproduct of urea cycle which causes neurotoxicity. Using DFMO (difluoromethylornithine) for inhibition of ODC1, they showed that ODC1 inhibition ameliorates behavioral impairments in an AD mouse model.
It would have been better if the authors had also investigated whether other cellular stressors—such as exposure to pathological tau, oxidative stress, and aging—would also influence the astrocytes in the same fashion.
The study also has many limitations, like use of DFMO as an ODC1 inhibitor (DFMO has been shown to have non-specific activities, side effects of DFMO, challenges in long-term inhibition of ODC1). Future studies are warranted to develop specific ODC1 inhibitors.
In conclusion, the study by Ju et al. provides a better understanding of the involvement of astrocyte-urea metabolism in AD pathophysiology. A deeper understanding of urea metabolism in reactive astrocytes may prove to be critical in understanding mechanisms of neurodegeneration.
References:
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017 Jan 26;541(7638):481-487. Epub 2017 Jan 18 PubMed.
Gaikwad S, Puangmalai N, Bittar A, Montalbano M, Garcia S, McAllen S, Bhatt N, Sonawane M, Sengupta U, Kayed R. Tau oligomer induced HMGB1 release contributes to cellular senescence and neuropathology linked to Alzheimer's disease and frontotemporal dementia. Cell Rep. 2021 Jul 20;36(3):109419. PubMed.
Cohen J, Torres C. Astrocyte senescence: Evidence and significance. Aging Cell. 2019 Jun;18(3):e12937. Epub 2019 Feb 27 PubMed.
Make a Comment
To make a comment you must login or register.