Lysosomes Take Center Stage in Parkinson’s and Frontotemporal Dementia
Quick Links
In neurodegenerative diseases, misfolded proteins pile up. Previous studies have pointed an accusing finger at faulty lysosomal garbage disposal systems and now, new evidence from several groups firmly entrenches this mechanism. It establishes a central role for lysosome function and lipid processing in the pathogenesis of Parkinson’s disease and a genetic form of frontotemporal dementia. The data call on drug developers to target lysosomes therapeutically, researchers said.
- Different Parkinson’s disease risk factors slot into the same pathogenic cascade.
- Knowledge from human neuron cultures can produce better mouse models of PD.
- Progranulin plays a role in lipid processing inside lysosomes.
In the case of Parkinson’s, familial mutations have implicated pathogenic processes including mitochondrial oxidative stress, excess α-synuclein, and lysosomal dysfunction. In the September 7 Science, scientists led by Dimitri Krainc at Northwestern University Feinberg School of Medicine, Chicago, attempted to unite all of these mechanisms in a single series of unfortunate events. Using induced neurons made from PD patients carrying mutations in the antioxidant gene DJ-1, the researchers found that oxidative stress led to the buildup of oxidized dopamine, which then dampened the efficiency of the lysosomal enzyme glucocerebrosidase (GBA). This, in turn, slowed lysosomal digestion, allowing α-synuclein to accumulate. “These pathways converge in a way that explains how different PD mutations can lead to the same disease,” Krainc told Alzforum.
Other papers offered complementary evidence for the importance of lysosomes. In the September 11 Nature Genetics, researchers led by Robert Graham and Tushar Bhangale at Genentech, South San Francisco, described the largest genetic meta-analysis of PD to date. Of the 17 new risk loci this work turned up, three candidate genes related to lysosomal function, continuing a pattern of enrichment for these among PD risk genes. Meanwhile, a September 6 paper in Brain from Anthony Schapira at University College London and colleagues, emphasized how a combination of pathogenic mutations in GBA and excess α-synuclein leads to toxicity.
“These papers … show clearly that lysosome dysfunction, going beyond GBA loss, is key to Parkinson’s pathogenesis,” John Hardy at UCL wrote to Alzforum (see full comment below).
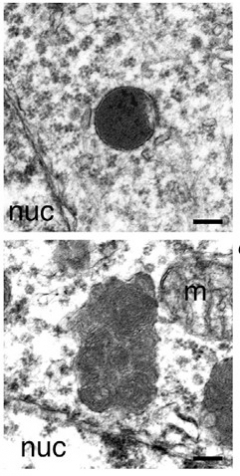
Lysosomal Indigestion.
Unlike smooth, round lysosomes in progranulin heterozygote mice (top), lysosomes in progranulin knockouts (bottom) accumulate undigested membrane, making them bumpy and swollen. [Courtesy of Cell Reports, Evers et al.]
What about frontotemporal dementia? On the face of it, this disorder has little in common with PD. FTD attacks behavior or language rather than movement, and features accumulation of tau or TDP-43 rather than α-synuclein. Dig a little deeper, however, and commonalities emerge, particularly with FTD cases caused by loss-of-function mutations in the growth factor progranulin. These account for up to 20 percent of familial and 5 percent of sporadic FTD. In the September 12 Cell Reports, researchers led by Joachim Herz at University of Texas Southwestern Medical Center, Dallas, reported that lysosomes lacking progranulin develop a bad case of indigestion. Chunks of membrane accumulate in these organelles, while the lipid composition of the cell shifts, suggesting problems with fat breakdown. Lo and behold, the changes are similar to those seen in lysosomal storage disorders, the authors note.
“This paper nicely adds to the mounting evidence that progranulin plays an important role in lysosomal function and homeostasis,” noted Thomas Kukar at Emory University, Atlanta (see full comment below).
A Web of Parkinson’s Mechanisms
Many studies have noted problems with lysosomes in PD (e.g., Jun 2012 news; Feb 2014 news; Jun 2014 news). Earlier work from Krainc and colleagues highlighted the connection between weak glucocerebrosidase activity and α-synuclein buildup (Jun 2011 news; Mar 2014 conference news). However, some PD mutations, such as DJ-1 variants, primarily affect mitochondrial oxidative stress (Nov 2002 news; Nov 2010 news).
To examine whether these pathways interrelate, Krainc and colleagues generated dopaminergic iPSC-derived neurons from two people with rare homozygous DJ-1 mutations, and compared them to neurons made from a person with a heterozygous DJ-1 mutation, as well as neurons from patients with idiopathic PD and controls. First author Lena Burbulla cultured these lines for months to track the development of pathology in them. The first problem to crop up was oxidative stress. By 50 days in vitro, the number of oxidized proteins spiked, while cellular respiration crashed. Next, the amount of oxidized dopamine surged 10-fold over control between days 70 and 150. Also starting at day 70, glucocerebrosidase activity slowed by 15 percent and α-synuclein levels increased fivefold. By six months, the disease process had reached lysosomes. Their efficiency dropped in half.
This pathogenic cascade was not unique to DJ-1 homozygotes. The heterozygous DJ-1 cell line also accumulated oxidized dopamine, though more slowly. Ditto for the idiopathic lines, which took up to six months to build up a three- to fivefold excess. GBA activity dropped at around the same time in these cells.
To see if there was a direct connection, the authors combined oxidized dopamine and GBA in cell-free experiments. The oxidized dopamine modified cysteine residues in GBA’s catalytic site, slashing the enzyme’s activity by two-thirds. Commenters appreciated this finding, and wondered if GBA from PD brain samples would show the same modification.
Although the time course data hinted at a linear cascade, Krainc noted that the full picture is more complex, with many steps running in both directions in toxic feedback loops. For example, high dopamine levels exacerbated mitochondrial oxidative stress. Other research has found that lysosomal dysfunction impairs the breakdown of malfunctioning mitochondria, and thus worsens oxidative stress (Gegg and Schapira, 2015). Likewise, high concentrations of α-synuclein can inhibit lysosomal digestion.
Because of these interrelationships, mutations at any point in this web could kick off Parkinson’s, Krainc suggested. In preliminary data, the authors found that induced neurons made from PD patients carrying mutations in parkin, PINK1, or LRRK2, or an extra copy of α-synuclein, also accumulated oxidized dopamine. Separately, the authors found that simply overexpressing α-synuclein in DJ-1 knockout mice boosted oxidized dopamine 10-fold and led to GBA dysfunction. The findings dovetail with Schapira’s Brain paper, in which he reported that mice with pathogenic GBA1 mutations did not lose dopaminergic neurons unless α-synuclein was overexpressed. The two factors together caused the neurons to die.
The importance of oxidized dopamine in pathogenesis may help explain why dopaminergic neurons of the substantia nigra are particularly vulnerable in Parkinson’s, noted Patrick Lewis at the University of Reading, U.K. “This addresses a fundamental question in neurodegeneration: Why is it that certain subsets of anatomically localized neurons die off?” Lewis believes induced human neurons may help researchers investigate this question for other neurodegenerative diseases, as well.
Man Versus Mouse
Studying human models is crucial because PD mice do not lose dopaminergic neurons. Not only that, dopaminergic neurons in wild-type mice do not form neuromelanin, the black deposit composed of oxidized dopamine mixed with other proteins and lipids. Krainc and colleagues found that cell lysates from the substantia nigra of DJ-1 knockout mice contained barely any oxidized dopamine, and their GBA activity and α-synuclein levels were normal. Mouse neurons appear to make less dopamine than human cells do, and seem to convert it into non-toxic metabolities rather than oxidized forms, Krainc noted. This could explain why PD mouse models retain healthy neurons.
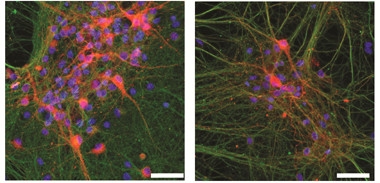
Toxic Formula.
Dopaminergic neurons (red) from DJ-1 knockout mice (left) thrive in culture until exposed to excess dopamine (right). [Courtesy of Science/AAAS.]
The authors wondered whether excess dopamine might make mouse dopaminergic neurons behave more like the human variety. They fed DJ-1 knockout mice L-DOPA, the dopamine precursor used to treat PD, for six months. The mice’s oxidized dopamine levels mushroomed sixfold, GBA activity dropped, and α-synuclein levels doubled. Voila, dopaminergic neurons began dying, to a 20 percent loss in the substantia nigra. The authors saw similar toxicity when they administered L-DOPA to DJ-1 knockout neurons in vitro (see image above). L-DOPA had no effect on wild-type neurons. Overall, the data suggest that excess dopamine combined with oxidative stress triggers Parkinson’s pathology, Krainc said.
Commenters found the strategy of feeding L-DOPA to DJ-1 knockout mice intriguing. The need for a better model is plain. “The Parkinson’s mouse models have been underwhelming,” noted Mark Cookson at the National Institutes of Health, Bethesda, Maryland. But does Krainc’s approach represent an improvement? It might seem counterintuitive given that in people, chronic dosing with L-DOPA is standard therapy. However, PD patients already have a massive loss of dopaminergic neurons and dopamine. Krainc’s data imply that early in disease, the problem may be too much dopamine. Cookson suggested that other labs try to replicate his data, and said he is interested in doing so himself. “If the finding holds up, this mouse model would be a big contribution to the field,” Cookson told Alzforum.
Researchers also noted that the findings validate the use of human cell models rather than rodents to dissect neurodegenerative disease pathways. “It’s important to study human neurons to identify targets for therapeutic development,” Krainc told Alzforum.
How About Treatment?
The therapeutic implications of the findings are less clear. Commenters noted that Parkinson’s disease attacks multiple brain systems and cell types, with glial dysfunction contributing as well, hence rescuing dopaminergic neurons alone might not treat all symptoms of PD (Jun 2017 news). Besides, Cookson noted, antioxidant therapy has repeatedly failed in Parkinson’s disease (May 2007 news; Mar 2014 news).
Nonetheless, Krainc believes that more specific and brain-penetrant antioxidants could hold potential. In his cell culture experiments, antioxidant treatment drove down accumulation of oxidized dopamine by three-fourths, preserving glucocerebrosidase and lysosomal function. As a result, α-synuclein levels in the neurons dropped by 80 percent. Inhibiting dopamine synthesis or blunting calcium signaling had similar benefits.
Another option is to unblock lysosomal function. In Krainc’s cell cultures, better lysosomal digestion reversed problems with glucocerebrosidase and lowered oxidized dopamine levels. Commenters agreed these data further strengthen the rationale for targeting lysosomes. Many groups are interested in this already. For example, Sanofi Genzyme is running a Phase 2 trial targeting a GBA substrate (Jul 2011 news; Dec 2016 conference news).

PD Gene Explosion. A meta-analysis nearly doubles the number of PD risk factors. New genes (red), known confirmed genes (black), and known genes not confirmed (gray) are shown by chromosome position. [Courtesy of Chang et al., Nature Genetics.]
Genetics Highlights Lysosomes. Again.
New genetic findings are fueling the lysosome fire. Graham and Bhangale first performed a GWAS with data from 6,476 people with Parkinson’s who were genotyped by the personal genomics company 23andMe, as per a collaborative agreement with Genentech (Jan 2015 news). First author Diana Chang combined this data with a previous PDGene meta-analysis and validated the findings in a third independent cohort, the NeuroX study (Jul 2014 news; Nalls et al., 2015). All told, the analysis included 26,035 cases and 403,190 controls, the largest to date.
This turned up 17 new risk loci for PD, while confirming many of the 24 known loci. Curiously, the authors found evidence that more than one PD-linked gene might be present in 10 of these 41 loci. Incidentally, adding the new loci to the known set improved the ability of a genetic risk score to detect Parkinson’s, with the area under the curve rising to 65 percent. By comparison, for Alzheimer’s this number stands at 84 percent, which makes it useful clinically (Escott-Price et al., 2017).
To find the genes flagged by the PD loci, the authors examined nearby candidates for functional variants. For 29 of the 41 loci, they nabbed either a protein-coding change or a variant that affected neuronal expression. In other cases, they could not pin down a functional variant, and several nearby genes remain candidates (see image above).
Importantly for this story, the candidate genes were enriched for associations with oxidation, autophagy, and lysosomal function. Two of the 17 new genes relate to mitochondria, three to lysosomes. The latter encode the protease cathepsin B, the proton pump ATPase H+ transporting V0 subunit a1 (ATP6V0A1), and the enzyme galactosylceramidase, which hydrolyzes lipids. Genentech researchers now are testing candidate genes in cellular assays to find out what they do.
Is Progranulin a Lysosomal Gene?
In contrast to these PD risk factors, the FTD gene progranulin has only recently been flagged as a lysosomal protein. Recent work shows that progranulin travels to lysosomes. There it is chopped into granulins, which remain there and may aid digestion (Sep 2015 news; Jul 2017 news; Aug 2017 news). As further evidence of this role, homozygous progranulin mutations cause a childhood-onset lysosomal storage disorder called neuronal ceroid lipofuscinosis (NCL), while heterozygous mutations trigger a surge in expression of lysosomal enzymes (Smith et al., 2012; Götzl et al., 2014).
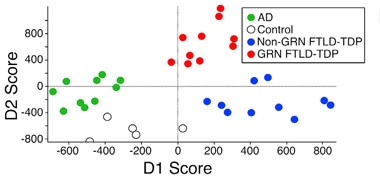
Fatty Fingerprints.
Brain samples from people with various neurodegenerative diseases contain distinct lipid profiles that can act as a disease signature. [Courtesy of Cell Reports, Evers et al.]
To dig into what progranulin and granulins do in lysosomes, joint first authors Bret Evers, Carlos Rodriguez-Navas, and Rachel Tesla first analyzed the lipid composition in postmortem cortical brain samples from people with autopsy-confirmed frontotemporal lobar degeneration (FTLD) due to progranulin mutations, FTLD from other causes, or Alzheimer’s disease, as well as healthy controls. Each group comprised samples from five people. Mass spectrometry revealed a unique lipid profile for each disorder, driven by changes in just a few classes of lipid (see image above).
Next, the authors turned to progranulin homozygote knockout mice. Their neurons contain more and larger lysosomes than neurons in heterozygote or control mice, and these hefty lysosomes are bulging with fragments of undigested cellular membrane like those seen in NCL (see electron micrograph at top of story). The authors examined lipid processing in embryonic fibroblasts from the progranulin knockout, heterozygote, and control mice. In knockouts and heterozygotes, they saw a gene dose-dependent accumulation of polyunsaturated triacylglycerides and a drop in diacylglycerides and the downstream metabolite phosphatidylserine. These are the same classes of lipid that are perturbed in the human brain samples. The findings suggest a role for granulins in snipping tri- into diacylglycerides, the authors noted.
As further evidence, the authors profiled gene expression in the knockout and control mice by microarray. In the knockouts, expression of hundreds of genes was altered, and they partially overlapped with those in NPC1 knockout mice. Lo and behold, effects were strongest on genes involved in lipid metabolism, lysosomal function, and immune response. The lysosomal genes were largely upregulated, perhaps as a compensatory response to impaired digestion, the authors suggested.
Other researchers were intrigued by the findings. “It is highly possible that this study reveals a basic physiologic function for progranulin and/or granulins in regulating lysosomal polyunsaturated fatty acid metabolism,” Chris Lee of the Biomedical Research Institute of New Jersey, Cedar Knolls, wrote to Alzforum (see full comment below). Li Gan at the Gladstone Institute of Neurological Disease, San Francisco, noted, “The effects on lysosomal lipid processing are very compelling, and offer a brand-new angle on how progranulin affects lipid metabolism.” Gan was particularly impressed by the lipid changes in progranulin heterozygote mouse cells, which typically do not have much of a phenotype. “This gene-dose effect supports the notion that NCL and FTD due to progranulin deficiency are essentially the same disease,” she added (see full comment below, and Ward et al., 2017).
Commenters agreed that the data overall strengthen the concept of Parkinson’s and FTD as lysosomal disorders. “In some sense, one can think of these diseases as formes frustes of lysosome storage diseases,” Hardy noted.—Madolyn Bowman Rogers
References
Alzpedia Citations
News Citations
- Evidence Piles Up for Lysosomal Dysfunction in Parkinson’s
- LRRK2 Interactions Identify New Parkinson’s Genes, Implicate Autophagy
- Gaucher’s Model Recapitulates Phenotype, Supports Drug Candidate
- Feedback Loop—Molecular Mechanism for PD, Gaucher’s Connection
- Protecting Neurons by Ramping Up Waste Disposal?
- New Parkinson’s Gene: DJ Mutations Make Neurons Change Their Tune
- Can Calcium Channel Blockers Save Stressed-Out Dopaminergic Neurons?
- Astrocytes, In Addition to Neural Networks, May Stoke Synuclein Pathology
- Coenzyme Q10 Fails to Quell Parkinson Symptoms
- Coenzyme Q10 Strikes Out in Phase 3 for Parkinson’s
- Targeting α-Synuclein, Glucocerebrosidase May Work for LBD
- Large Phase 2 Trial Starting Up in Genetic Parkinson’s Population
- Genentech Strikes Deal with 23andMe to Study Parkinson’s Genomes
- Largest Meta-GWAS Yet Uncovers New Genetic Links to Parkinson’s
- Rare FTD Cases Another Example of Lysosomal Pathology in Dementia
- TMEM106B and Progranulin Duke It Out at the Lysosome
- Granulins: The Missing Link between Progranulin and Lysosome Function?
Paper Citations
- Gegg ME, Schapira AH. Mitochondrial dysfunction associated with glucocerebrosidase deficiency. Neurobiol Dis. 2015 Sep 24; PubMed.
- Nalls MA, Bras J, Hernandez DG, Keller MF, Majounie E, Renton AE, Saad M, Jansen I, Guerreiro R, Lubbe S, Plagnol V, Gibbs JR, Schulte C, Pankratz N, Sutherland M, Bertram L, Lill CM, DeStefano AL, Faroud T, Eriksson N, Tung JY, Edsall C, Nichols N, Brooks J, Arepalli S, Pliner H, Letson C, Heutink P, Martinez M, Gasser T, Traynor BJ, Wood N, Hardy J, Singleton AB, International Parkinson's Disease Genomics Consortium (IPDGC), Parkinson's Disease meta-analysis consortium. NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol Aging. 2015 Mar;36(3):1605.e7-12. Epub 2014 Aug 4 PubMed.
- Escott-Price V, Myers AJ, Huentelman M, Hardy J. Polygenic risk score analysis of pathologically confirmed Alzheimer disease. Ann Neurol. 2017 Aug;82(2):311-314. Epub 2017 Aug 9 PubMed.
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, Sims KB, Lewis J, Lin WL, Dickson DW, Dahl HH, Bahlo M, Berkovic SF. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012 Jun 8;90(6):1102-7. PubMed.
- Götzl JK, Mori K, Damme M, Fellerer K, Tahirovic S, Kleinberger G, Janssens J, van der Zee J, Lang CM, Kremmer E, Martin JJ, Engelborghs S, Kretzschmar HA, Arzberger T, Van Broeckhoven C, Haass C, Capell A. Common pathobiochemical hallmarks of progranulin-associated frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis. Acta Neuropathol. 2014 Mar 12; PubMed.
- Ward ME, Chen R, Huang HY, Ludwig C, Telpoukhovskaia M, Taubes A, Boudin H, Minami SS, Reichert M, Albrecht P, Gelfand JM, Cruz-Herranz A, Cordano C, Alavi MV, Leslie S, Seeley WW, Miller BL, Bigio E, Mesulam MM, Bogyo MS, Mackenzie IR, Staropoli JF, Cotman SL, Huang EJ, Gan L, Green AJ. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med. 2017 Apr 12;9(385) PubMed.
External Citations
Further Reading
Primary Papers
- Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, Santos DP, Blanz J, Obermaier CD, Strojny C, Savas JN, Kiskinis E, Zhuang X, Krüger R, Surmeier DJ, Krainc D. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science. 2017 Sep 22;357(6357):1255-1261. Epub 2017 Sep 7 PubMed.
- Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F, International Parkinson's Disease Genomics Consortium, 23andMe Research Team, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet. 2017 Sep 11; PubMed.
- Evers BM, Rodriguez-Navas C, Tesla RJ, Prange-Kiel J, Wasser CR, Yoo KS, McDonald J, Cenik B, Ravenscroft TA, Plattner F, Rademakers R, Yu G, White CL 3rd, Herz J. Lipidomic and Transcriptomic Basis of Lysosomal Dysfunction in Progranulin Deficiency. Cell Rep. 2017 Sep 12;20(11):2565-2574. PubMed.
- Migdalska-Richards A, Wegrzynowicz M, Rusconi R, Deangeli G, Di Monte DA, Spillantini MG, Schapira AH. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain. 2017 Oct 1;140(10):2706-2721. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Biomedical Research Institute of New Jersey / Atlantic Health System
The finding in Evers et al. strongly supports that PGRN/GRN is a key regulator in lysosomal polyunsaturated fatty acid metabolism (PUFA).
Because the lipidomic profile changes under PGRN-deficient conditions were consistent in both human brain tissues, i.e., disease end-stage, and in mouse embryonic fibroblasts, i.e., physiologically normal, it is highly possible that the study reveals a basic physiologic function for PGRN/GRN.
Given the recent reports on PGRN processing and the presence of mature GRN protein in the lysosomes (Lee et al., 2017; Holler et al., 2017; Zhou et al., 2017), the current study has narrowed the search of GRN protein targets to the metabolic pathways. Mapping out the lysosomal GRN pathway will further improve our understanding of the disease mechanism of PGRN-related disorders.
References:
Lee CW, Stankowski JN, Chew J, Cook CN, Lam YW, Almeida S, Carlomagno Y, Lau KF, Prudencio M, Gao FB, Bogyo M, Dickson DW, Petrucelli L. The lysosomal protein cathepsin L is a progranulin protease. Mol Neurodegener. 2017 Jul 25;12(1):55. PubMed.
Holler CJ, Taylor G, Deng Q, Kukar T. Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins that Are Haploinsufficient in Patients with Frontotemporal Dementia Caused byMutations. eNeuro. 2017 Jul-Aug;4(4) Epub 2017 Aug 18 PubMed.
Zhou X, Paushter DH, Feng T, Sun L, Reinheckel T, Hu F. Lysosomal processing of progranulin. Mol Neurodegener. 2017 Aug 23;12(1):62. PubMed.
View all comments by Chris LeeUniversity of California, San Francisco
There are two exciting aspects of this paper.
One is the finding that progranulin deficiency (full-length and granulins) leads to gene-dose alterations in lipid metabolism. The effects on lysosomal lipid processing are very compelling, and offer a brand-new angle on how progranulin affects lipid metabolism. This is an under-studied field that could be of enormous importance in aging and neurodegeneration. The big unanswered question is how PGRN affects lipid metabolism, and what are the downstream effects in different cell types? How is this related to the altered inflammatory phenotype in innate immune cells?
Secondly, the gene dose-dependent effects reported here are unprecedented in mouse cells. In many previous studies, Grn+/– mouse cells have little if any detectable phenotype. The gene dose effect reported here supports the notion that NCL and FTD due to progranulin deficiency are essentially the same disease with shared mechanism (also see Ward et al., 2017). The authors also offered additional evidence with the comparison with the transcriptomes from NPC cells.
From a therapeutic point, this study, along with others, puts targeting lysosomal dysfunction at the center of pharmacologic intervention.
References:
Ward ME, Chen R, Huang HY, Ludwig C, Telpoukhovskaia M, Taubes A, Boudin H, Minami SS, Reichert M, Albrecht P, Gelfand JM, Cruz-Herranz A, Cordano C, Alavi MV, Leslie S, Seeley WW, Miller BL, Bigio E, Mesulam MM, Bogyo MS, Mackenzie IR, Staropoli JF, Cotman SL, Huang EJ, Gan L, Green AJ. Individuals with progranulin haploinsufficiency exhibit features of neuronal ceroid lipofuscinosis. Sci Transl Med. 2017 Apr 12;9(385) PubMed.
View all comments by Li GanEmory University
Emory University
This new paper from Joachim Herz’s lab nicely adds to the mounting evidence that progranulin (PGRN) has an important role in lysosomal function and homeostasis.
Lysosome dysfunction, autophagy impairment, and alterations in lipid metabolism are found in Alzheimer’s disease, frontotemporal dementia, and many other neurodegenerative disorders. Here the authors uncover that complete or partial loss of PGRN in both humans and mice leads to changes in specific lipid profiles.
However, the mechanism whereby decreased or absent PGRN causes lysosome impairment and lipid alterations is unclear. We have recently found that PGRN is rapidly processed into stable granulin proteins (GRNs) in the lysosome, which are haploinsufficient in FTD-GRN patients (Holler et al., 2017). One potential explanation that accounts for both observations is that loss of lysosomal granulins leads to defects in lipid turnover. More exciting work lies ahead to uncover the intricacies of the function of PGRN and GRNs in the lysosome in order to develop therapies where decreased levels of PGRN/GRNs are implicated.
References:
Holler CJ, Taylor G, Deng Q, Kukar T. Intracellular Proteolysis of Progranulin Generates Stable, Lysosomal Granulins that Are Haploinsufficient in Patients with Frontotemporal Dementia Caused byMutations. eNeuro. 2017 Jul-Aug;4(4) Epub 2017 Aug 18 PubMed.
View all comments by Christopher HollerInstitute of Neurology, UCL
The connection between lysosome dysfunction and Parkinson’s disease is now clear. These papers—as well as one this week by Schapira in Brain and another due out soon from Shulman, Heutink, and the IPDGC—show clearly that lysosome dysfunction, going beyond GBA loss, is key to Parkinson’s pathogenesis. This is indeed an exciting insight.
It is also the case that most pure frontotemporal dementias are caused by different variants in the endosomal/lysosome system. In some sense, one can think of these diseases as formes frustes of lysosome storage diseases.
Less easy to understand is the idea of a “special relationship” between lysosomal storage diseases and dopamine neurons, since we now know there is nothing special about DA neurons in PD. Braaks’ staging and other work shows that the substantia nigra neurons are but one type of neuron hit by the PD process. They are not the first, and the process is by no means selective.
References:
Migdalska-Richards A, Wegrzynowicz M, Rusconi R, Deangeli G, Di Monte DA, Spillantini MG, Schapira AH. The L444P Gba1 mutation enhances alpha-synuclein induced loss of nigral dopaminergic neurons in mice. Brain, 21 Sept. 2017
Make a Comment
To make a comment you must login or register.