Cholesterol Trafficking Takes a Hit in Alzheimer’s Neurons
Quick Links
Numerous studies have found defects in endocytosis in Alzheimer’s disease. A paper in the October 11 Cell Reports adds a new twist, reporting that induced human neurons carrying familial AD mutations poorly take up amyloid precursor protein (APP) and lipoproteins, and fail to sort these molecules properly for transport to axons. A dearth of axonal lipoprotein could starve synapses, which depend on cholesterol for proper functioning, noted Lawrence Goldstein at the University of California, San Diego, who led the research. Exactly how AD mutations impair endocytosis and transcytosis remains unclear. However, inhibiting the secretase BACE, which cleaves APP to produce the β-CTF fragment, rescued the defect. β-CTF may play a role in trapping proteins in endosomes, Goldstein suggested. The data strengthen previous findings that this fragment can have toxic effects. The research is also one of the first to study the consequences of engineering FAD mutations into neurons derived from normal iPS cells.
Other researchers called the data intriguing. “This is a great effort to look at transport using human induced pluripotent stem cell-derived neurons, and the authors find something that no one has reported using mouse models or human brain samples,” said Gopal Thinakaran at the University of Chicago. He noted, however, that the mechanisms behind this finding need to be elucidated. Ralph Nixon at the Nathan Kline Institute in Orangeburg, New York, agreed, pointing out that many previous studies have reported accelerated, rather than impaired, endocytosis in Alzheimer’s neurons. Commenters said it will be important to replicate these findings in other model systems, including animals.
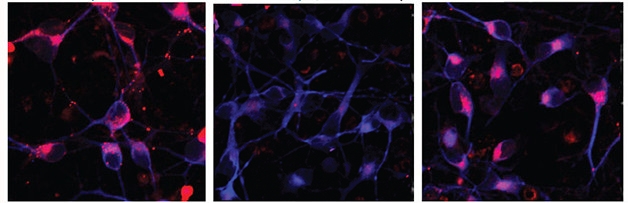
Rescuing Endocytosis. Induced human neurons (blue) carrying a presenilin mutation (middle) take up little cholesterol (red) compared to wild-types (left). Inhibiting BACE1 restores uptake (right). [Courtesy of Cell Reports, Woodruff et al.]
Earlier studies by Nixon and others found abnormal endocytosis in AD brains as well as in models of Down’s syndrome (see Cataldo et al., 2001; Ginsberg et al., 2010; Jul 2006 news). Cellular and genetic studies likewise suggest a key role for endocytosis in Alzheimer’s pathogenesis (see Oct 2011 news). Moreover, Nixon previously implicated β-CTF as a cause of these endocytic problems (see Jan 2010 news; Jul 2015 news).
These cellular studies, however, relied on APP overexpression. Goldstein and colleagues wanted to look at isolated neurons that expressed endogenous levels of protein. To do this, the researchers generated human neurons from iPS cells, and used gene editing to introduce the ΔE9 familial mutation in presenilin 1 (see Jan 2012 news; Woodruff et al., 2013). Joint first authors Grace Woodruff and Sol Reyna then measured how well these neurons internalized various molecules. The cells took up about 25 percent less APP and low-density lipoprotein (LDL) than control neurons did, but normal amounts of other proteins and sugars. In addition to these changes in endocytosis, slightly less endogenous APP reached the axons than in control neurons, and about half as much LDL, indicating impaired vesicle sorting and transport to axons that might particularly affect cholesterol dynamics.
What might explain the anemic uptake and transport of these proteins? For clues, the authors looked to the GTPase Rab11, which facilitates endosome recycling and axonal transcytosis of cargo such as BACE1 and APP (see Buggia-Prévot et al., 2014; Niederst et al., 2015). In the presenilin mutant neurons, about 25 percent less Rab11 reached axons from the cell body than in wild-type cells, suggesting impaired trafficking. To further explore its potential role in APP/LDL transport, the authors knocked down Rab11 in wild-type neurons. This completely abolished endocytosis and transcytosis of LDL, while cutting axonal APP by 40 percent. In ongoing work, the authors are overexpressing Rab11 in the mutant neurons to see if that will rescue trafficking.
These experiments did not answer how FAD mutations might cause problems with Rab11 and endosomal trafficking. Goldstein believes there may be direct and indirect effects. He pointed out that presenilin has been found to physically interact with Rab11 in vesicle membranes, though he has not tested this himself, and that the ΔE9 PS mutation causes a buildup of β-CTF by impairing its cleavage by γ-secretase (see Dumanchin et al., 1999). To see if β-CTFs impede transport, the authors treated wild-type neurons with a γ-secretase inhibitor, which pumped up levels of the fragment. This suppressed endocytosis of LDL just as the ΔE9 mutation did. When they treated mutant neurons with a BACE inhibitor to prevent production of β-CTF, LDL trafficking rebounded to normal (see image above). The evidence supports a toxic effect of β-CTF on endocytosis, Goldstein said.
Intriguingly, some previous papers also reported that endosome recycling and lipoprotein uptake required functional presenilin (see Zhang et al., 2006; Tamboli et al., 2008). William Mobley at UCSD concurs that β-CTF harms endocytosis and trafficking, although how it does so is still unknown. “The findings are really interesting, and certainly will support the field’s need to consider vesicle trafficking in formulations of pathogenesis,” he wrote to Alzforum.
The authors then extended the research to two APP variants that cause familial AD, the Swedish and Indiana mutations. Just like the neurons expressing presenilin mutants, induced neurons carrying these APP mutations internalized less LDL than did wild-type cells. In keeping with the idea that β-CTF might suppress endocytosis, BACE inhibition rescued uptake of LDL in Indiana mutant cells. However, BACE inhibitors had no effect on endocytosis in the APPSw neurons. APPSw gets rapidly cleaved by BACE, and Goldstein noted inhibitors have little effect on its processing (see Yamakawa et al., 2010), though others were not so sure. “We think both APP and PS mutants affect the recycling endosome in the same way,” Goldstein told Alzforum.
Goldstein wonders if impaired lipoprotein uptake and transcytosis might exacerbate sporadic Alzheimer’s disease. There are hints of this already. ApoE transports cholesterol, and the E4 variant, which is believed to poorly ferry lipids, boosts Alzheimer’s risk as much as 12-fold when homozygous. “Defective transcytosis may link familial and sporadic forms of Alzheimer’s disease,” he speculated. If so, BACE inhibitors may prove to be an even more potent therapy than currently expected, Goldstein noted. He suggested that targeting endocytic recycling and sorting might be a fruitful therapeutic path, as well.
Commenters noted that this study raises many new questions. Thinakaran found it puzzling that the endocytosis defect affected the transcytosis of LDL more than that of APP, when both molecules interact with endocytic machinery through an identical motif. He also wondered if β-CTF was really the culprit in the transport problems, given that BACE inhibition did not rescue both Swedish and Indiana APP mutants. Nixon stressed the importance of looking at endocytosis and transcytosis in other presenilin variants, given that the ΔE9 mutation produces atypical “cotton-wool” plaques in the brain rather than classic neuritic plaques. Others were intrigued by the potential downstream effects. “In the future, it will be interesting to determine the impact of [reduced] neuronal transcytosis on synaptic activity,” suggested Claudia Almeida at NOVA Medical School, Lisbon, Portugal, in an email to Alzforum.—Madolyn Bowman Rogers
References
News Citations
- Trisomy Trouble: Neurotrophin Signaling Defective in Down Syndrome
- Traffic Control—Curb Endocytosis to Curb AD Pathogenesis?
- APP in Pieces: βCTF implicated in Endosome Dysfunction
- Partners in Crime: APP Fragment and Endosomal Protein Impair Endocytosis
- Induced Neurons From AD Patients Hint at Disease Mechanisms
Mutations Citations
Paper Citations
- Cataldo A, Rebeck GW, Ghetri B, Hulette C, Lippa C, Van Broeckhoven C, van Duijn C, Cras P, Bogdanovic N, Bird T, Peterhoff C, Nixon R. Endocytic disturbances distinguish among subtypes of Alzheimer's disease and related disorders. Ann Neurol. 2001 Nov;50(5):661-5. PubMed.
- Ginsberg SD, Alldred MJ, Counts SE, Cataldo AM, Neve RL, Jiang Y, Wuu J, Chao MV, Mufson EJ, Nixon RA, Che S. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biol Psychiatry. 2010 Nov 15;68(10):885-93. Epub 2010 Jul 23 PubMed.
- Woodruff G, Young JE, Martinez FJ, Buen F, Gore A, Kinaga J, Li Z, Yuan SH, Zhang K, Goldstein LS. The presenilin-1 ΔE9 mutation results in reduced γ-secretase activity, but not total loss of PS1 function, in isogenic human stem cells. Cell Rep. 2013 Nov 27;5(4):974-85. Epub 2013 Nov 14 PubMed.
- Buggia-Prévot V, Fernandez CG, Riordan S, Vetrivel KS, Roseman J, Waters J, Bindokas VP, Vassar R, Thinakaran G. Axonal BACE1 dynamics and targeting in hippocampal neurons: a role for Rab11 GTPase. Mol Neurodegener. 2014 Jan 4;9:1. PubMed.
- Niederst ED, Reyna SM, Goldstein LS. Axonal amyloid precursor protein and its fragments undergo somatodendritic endocytosis and processing. Mol Biol Cell. 2015 Jan 15;26(2):205-17. Epub 2014 Nov 12 PubMed.
- Dumanchin C, Czech C, Campion D, Cuif MH, Poyot T, Martin C, Charbonnier F, Goud B, Pradier L, Frebourg T. Presenilins interact with Rab11, a small GTPase involved in the regulation of vesicular transport. Hum Mol Genet. 1999 Jul;8(7):1263-9. PubMed.
- Zhang M, Haapasalo A, Kim DY, Ingano LA, Pettingell WH, Kovacs DM. Presenilin/gamma-secretase activity regulates protein clearance from the endocytic recycling compartment. FASEB J. 2006 Jun;20(8):1176-8. PubMed.
- Tamboli IY, Prager K, Thal DR, Thelen KM, Dewachter I, Pietrzik CU, St George-Hyslop P, Sisodia SS, De Strooper B, Heneka MT, Filippov MA, Müller U, Van Leuven F, Lütjohann D, Walter J. Loss of gamma-secretase function impairs endocytosis of lipoprotein particles and membrane cholesterol homeostasis. J Neurosci. 2008 Nov 12;28(46):12097-106. PubMed.
- Yamakawa H, Yagishita S, Futai E, Ishiura S. beta-Secretase inhibitor potency is decreased by aberrant beta-cleavage location of the "Swedish mutant" amyloid precursor protein. J Biol Chem. 2010 Jan 15;285(3):1634-42. PubMed.
Further Reading
Primary Papers
- Woodruff G, Reyna SM, Dunlap M, Van Der Kant R, Callender JA, Young JE, Roberts EA, Goldstein LS. Defective Transcytosis of APP and Lipoproteins in Human iPSC-Derived Neurons with Familial Alzheimer's Disease Mutations. Cell Rep. 2016 Oct 11;17(3):759-773. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Arkansas for Medical Sciences
This study by Woodruff et al. has provided intriguing new ideas about the roles of enzymes that have previously garnered interest almost exclusively due to their effects on APP. BACE1 and γ-secretase are well-known to have effects on other substrates, and this work ties at least some of those substrates—the lipoprotein receptor family—to these enzymes via the functional interactions between these receptors and APP.
However, there are direct physical interactions between lipoprotein receptors and γ-secretase, too. It has been known for some time that certain lipoprotein receptors are substrates for γ-secretase, and the effects of presenilin mutation or γ-secretase inhibitors on LDL uptake are compelling. Until recently, however, no effect of presenilin mutation on the direct processing of lipoprotein receptors had been documented.
We have now shown (Wang et al., 2016) that three mutations of presenilin-1 impair the processing and/or trafficking of LRP8, a member of the lipoprotein receptor family that is somewhat unusual for the signal transduction pathways it activates in response to Reelin and perhaps other ligands. The effects we uncovered are understandably concentrated on the further processing of the C-terminal "stump" created by sheddases, particularly after Reelin application. Thus, biological consequences may derive primarily from impacts on the gene-regulatory function of the "LICD" (LRP8 C-terminal domain). Nevertheless, we also found changes in the full-length LRP8 in cells expressing mutated presenilin-1. Thus, familial AD mutations in presenilin-1 may dysregulate lipoprotein receptor function, including internalization of lipoprotein particles, due to the status of these receptors as substrates of γ-secretase.
References:
Wang W, Moerman-Herzog AM, Slaton A, Barger SW. Presenilin 1 mutations influence processing and trafficking of the ApoE receptor apoER2. Neurobiol Aging. 2017 Jan;49:145-153. Epub 2016 Oct 11 PubMed.
Make a Comment
To make a comment you must login or register.