Partners in Crime: APP Fragment and Endosomal Protein Impair Endocytosis
Quick Links
In Alzheimer’s disease, enlarged endosomes hamper waste disposal pathways in neurons. In the July 21 Molecular Psychiatry, researchers led by Ralph Nixon at the Nathan Kline Institute in Orangeburg, New York, extend previous research that implicated the β-C-terminal fragment of amyloid precursor protein in this process. The researchers drilled down into the mechanism in cell culture models, finding that excess β-CTF recruits a protein called APPL1 to early endosomes. APPL1 helps rev up endocytosis, causing vesicles to swell and get stuck as they travel along axons. Knockdown of APPL1 reversed these effects, suggesting this mechanism could be targeted therapeutically. The findings shed light on one of the earliest pathologic features of Alzheimer’s, say the authors.
Commenters found the data intriguing. Greg Cole at the University of California, Los Angeles, wrote, “This is a novel mechanism and potentially very important … it seems likely to offer new therapeutic targets.” (See full comment below.) Stefan Lichtenthaler at the German Center for Neurodegenerative Diseases, Bonn, noted that the finding helps elucidate the role of β-CTF in toxicity. “We long thought that β-CTF may have an ‘unspecific’ neurotoxic function when it accumulates, but now this paper shows a specific mechanism,” he wrote to Alzforum. He noted that in general, BACE-derived CTFs may not just be fragments on the path of membrane protein degradation, but have specific physiological roles (see full comment below).
Previous studies by Nixon and others suggest that neuronal endocytic and lysosomal systems become gummed up early in Alzheimer’s disease. Endocytosis speeds up, lysosomal degradation can't keep pace and grinds to a halt, and endosomal vesicles become swollen and accumulate in neurites (see Feb 2005 news; Jun 2010 news; May 2011 news). The small GTPase rab5, which switches on in AD, regulates endosome trafficking and drives these changes (see Stenmark, 2009; Cataldo et al., 2008). Nixon reported that the β-CTF fragment of APP also contributed, but it was not clear how (see Jan 2010 news).
Since β-CTF did not directly bind rab5, Nixon and colleagues looked for molecules that might mediate their interaction. They focused on APPL1, aka adaptor protein containing pleckstrin homology domain, phosphotyrosine-binding (PTB) domain, and leucine zipper motif. Despite its acronym, this protein is not related to APP or to amyloid precursor-like protein 1 (APLP1). As an adaptor protein, APPL1 brings other proteins together. It binds and stabilizes active rab5 on early endosomes inside the cell (see Miaczynska et al., 2004). Because amyloid precursor protein is known to bind PTB domains, the authors considered the β-CTF cytosolic domain of APP a good candidate to interact with APPL1 as well (see Tamayev et al., 2009).
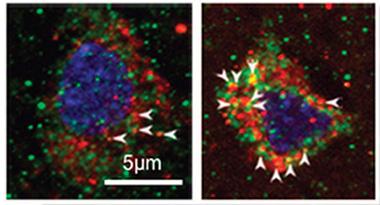
Activated Endosomes.
In AD brain (right), endosomes containing both APPL1 (green) and rab5 (red) abound compared to control brain (left). [Courtesy of Kim et al., Molecular Psychiatry.]
Using co-immunoprecipitations, first author Seonil Kim found that β-CTF bound directly to APPL1, and that he could abolish this by mutating the interaction domains on either protein. To investigate the consequences of binding, the authors turned to a neuroblastoma cell line that overexpressed human wild-type APP. Because APP is processed in endosomes, excessive β-CTF should accumulate there, allowing APPL1 to be recruited in turn. Consistent with this, the authors saw more APPL1 decorating early endosomes in these cells than in a control neuroblastoma line. Moreover, the APP line had oversized vesicles, 50 percent more activated rab5, and twofold more endocytosis overall. Knockdown of APPL1 shrank vesicles back to normal, lowered active rab5, and slowed endocytosis.
In Nixon’s 2010 study, he found that BACE inhibitors, which lower β-CTF levels, restored normal endocytosis, while γ-secretase inhibitors, which pump up β-CTF levels, worsened it. Those studies were done in fibroblasts from people with Down’s syndrome, who carry an extra copy of APP and develop Alzheimer’s-like pathology in middle age. Evidence suggests that endosomes malfunction decades earlier in DS (see Cataldo et al., 2000). The authors wanted to see if APPL1 mediated β-CTF’s effects in DS endosomes as well. Consistent with this, they saw more APPL1 on enlarged endosomes in DS fibroblasts compared to controls. Knockdown of APPL1 restored vesicles to normal size.
Endosomal dysfunction has numerous downstream effects, including slowing endosome transport along axons. Would APPL1 mediate these also? Watching vesicle movements in mouse cortical neuron cultures via time-lapse photography, the authors saw that in cells overexpressing wild-type APP and rab5, endosomes crawled along axons and made frequent stops, whereas in control cells they moved quickly and steadily. The largest endosomes, those with a surface area greater than 0.5 square microns, halted most frequently. These big vesicles may get stuck trying to push through cell cytoplasm, the authors suggested. Again, knockdown of APPL1 shrank endosomes and completely reversed these transport defects.
Endosomes also act as platforms for cell signaling, with rab5 activating numerous downstream molecules. These include NF-κB, which regulates inflammation and apoptosis and is implicated in both Alzheimer’s disease and Down’s syndrome (see Engidawork et al., 2001; Tilstra et al., 2011). In Down’s syndrome cells, levels of a downstream activator of NF-κB signaling were up 75 percent in the nucleus compared with control fibroblast levels. Knockdown of APPL1 restored levels to normal, again demonstrating the key role of APPL1 in endosome dysfunction.
While these cell culture results implicate APPL1 in numerous Alzheimer’s pathologies, does any of this translate to human brain? Preliminary results suggest they might. The authors found elevated β-CTF in 13 postmortem AD brains compared to 13 controls, as might be expected since BACE activity has been found to be high in AD (see Sep 2002 news). AD patients also had twofold more APPL1 on enlarged endosomes in cortical neurons than controls (see image above). Other studies have also reported abnormal distribution of APPL1 in AD brains (see Ogawa et al., 2013).
Numerous questions remain. In future work, Nixon will examine the effects of APPL1 on downstream late endosome and lysosome function, and whether this contributes to the failure of autophagy in AD. Because ApoE4 and cholesterol also activate rab5, and cholesterol binds β-CTF, he will investigate whether ApoE4 and cholesterol play a role in endosome regulation as well. One possibility is that ApoE4 and cholesterol might increase the concentration of β-CTF in endosomes, said Nixon.
The results suggest that lowering APPL1 or β-CTF might be therapeutic. Knockdown of APPL1 did not harm normal endocytosis in cultured cells, hinting that it might be safe to target this protein, Nixon noted. Commentators pointed out that BACE inhibitors might provide a double benefit to AD patients because they suppress production of both Aβ and β-CTF. “We can hope BACE inhibitors may have some therapeutic benefits that are not shared by γ-secretase inhibitors or most Aβ immunotherapy approaches,” Cole wrote to Alzforum. Several such inhibitors are currently in clinical trials (see Dec 2013 news; Oct 2014 news; Apr 2015 conference news).—Madolyn Bowman Rogers
References
News Citations
- Varicose Axons: Traffic Jams Precede AD Pathology in Mice, Men
- Death of the Neatnik: Neurons Perish When Trash Clutters Their Space?
- Lysosomal Block Clogs Transport, Swells Neurites
- APP in Pieces: βCTF implicated in Endosome Dysfunction
- BACE Above Base in Alzheimer’s Patients
- Merck BACE Inhibitor Clears a Safety Hurdle, Gets New Trial
- Lilly Teams Up With AstraZeneca for BACE Inhibitor Phase 2/3 Trial
- At AD/PD Meeting, New BACE Inhibitor Struts Its Stuff
Paper Citations
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009 Aug;10(8):513-25. Epub 2009 Jul 15 PubMed.
- Cataldo AM, Mathews PM, Boiteau AB, Hassinger LC, Peterhoff CM, Jiang Y, Mullaney K, Neve RL, Gruenberg J, Nixon RA. Down syndrome fibroblast model of Alzheimer-related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am J Pathol. 2008 Aug;173(2):370-84. PubMed.
- Miaczynska M, Christoforidis S, Giner A, Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG, Zerial M. APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment. Cell. 2004 Feb 6;116(3):445-56. PubMed.
- Tamayev R, Zhou D, D'Adamio L. The interactome of the amyloid beta precursor protein family members is shaped by phosphorylation of their intracellular domains. Mol Neurodegener. 2009;4:28. PubMed.
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000 Jul;157(1):277-86. PubMed.
- Engidawork E, Gulesserian T, Seidl R, Cairns N, Lubec G. Expression of apoptosis related proteins: RAIDD, ZIP kinase, Bim/BOD, p21, Bax, Bcl-2 and NF-kappaB in brains of patients with Down syndrome. J Neural Transm Suppl. 2001;(61):181-92. PubMed.
- Tilstra JS, Clauson CL, Niedernhofer LJ, Robbins PD. NF-κB in Aging and Disease. Aging Dis. 2011 Dec;2(6):449-65. PubMed.
- Ogawa A, Yamazaki Y, Nakamori M, Takahashi T, Kurashige T, Hiji M, Nagano Y, Yamawaki T, Matsumoto M. Characterization and distribution of adaptor protein containing a PH domain, PTB domain and leucine zipper motif (APPL1) in Alzheimer's disease hippocampus: an immunohistochemical study. Brain Res. 2013 Feb 4;1494:118-24. PubMed.
Further Reading
Primary Papers
- Kim S, Sato Y, Mohan PS, Peterhoff C, Pensalfini A, Rigoglioso A, Jiang Y, Nixon RA. Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer's disease. Mol Psychiatry. 2015 Jul 21; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
German Center for Neurodegenerative Diseases (DZNE)
This is an interesting paper, which follows up on an earlier paper in PNAS in 2010 from the same group. Randy has been following the endocytic connection in AD for many years and has published very well on the topic. The paper provides additional evidence for a selective role of the β-CTF of APP (C99). We long thought that β-CTF may have an "unspecific" neurotoxic function when it accumulates, but this paper shows a specific mechanism, namely the recruitment of APPL1 and the activation of Rab5, two endocytic proteins. Thus, when we speak about roles of APP and its fragments, we should not only speak about full-length APP and Aβ, but also about β-CTF. A recent study from Bart De Strooper’s lab (Barao et al., Cell Reports, in press) demonstrates that the β-CTF of another protein, the BACE1 substrate CHL1, is required for growth cone collapse in thalamic neurons. Together these studies raise the intriguing possibility that—more generally—β-CTFs may not just be fragments on the path of membrane protein degradation, but may have specific physiological roles.
APPL1 should not be confused with the APP homolog APLP1, which is an unrelated protein. The APPL1 knock-down and rescue experiments are very convincing, in particular in the DS fibroblasts. Future experiments should further investigate the direct interaction between β-CTF and APPL1 when both proteins are expressed under endogenous conditions (and not just overexpressed as done here), although this may be technically challenging. Additionally, knockdown/knockout experiments in AD models, including AD mice, may be very helpful to further demonstrate a role for β-CTF not only in DS, but also in AD.
UCLA/VA
I found this paper both convincing and very interesting because: 1) The in vitro results provide a specific mechanism for an APP-dependent but Aβ-independent toxicity pathway via β-CTF recruitment of Rab5 and APPL1 to the endosomal\lysosomal system. 2) This APPL1 pathway is upstream from NF-κB activation in neurons and potentially also in glia with many downstream consequences. 3) γ-Secretase inhibition increased β-CTF and made endosomal/lysosomal pathology worse, which suggests this pathway might help explain some of the adverse effects of γ-secretase inhibition. 4) The authors show increases in β-CTF and APPL1 in AD and the APPLI co-localizes with Rab5—arguing that this pathway is activated in AD.
This is a novel mechanism and potentially very important. While it is difficult to predict the net contribution of this new pathway to other AD pathologies and symptoms, it seems likely to play some significant role and it offers new therapeutic targets. In addition, the potential correction of this new independent role for β-CTF in causing endosomal pathology is another reason we can hope BACE inhibitors may have some therapeutic benefits that are not shared by γ-secretase inhibitors or most Aβ immunotherapy approaches.
Incidentally, the curry spice component curcumin is an NF-κB inhibitor. Since we have developed and been pursuing a more bioavailable form of curcumin for clinical trials with AD, we have encountered a community of parents of children with Down’s syndrome who have been treating their children with curcumin and blogging about improvements in their symptoms. Similar anecdotal reports suggest possible benefits in AD. Therefore, it is intriguing to see this new pathway as an upstream activator of NF-κB in AD and DS.
University of Zurich
The paper is an elegant piece of work demonstrating, for the first time, the molecular basis of how b-CTF could play a role in the activation of Rab5, a process that is intricately associated with the enlargement of endosomes. Apart from the plaques and tangles, endocytic abnormalities are also cardinal features of AD. Nixon and co-workers now conclusively demonstrate that b-CTF binds to the Rab5 effector, APPL1, and thereby affects the activation of Rab5. This is, to my best of the knowledge, the first time, a molecular mechanism that links APP processing and enlargement of the endosomes has been identified and hence I am very enthusiastic about this work. Back in 2006 we identified Rab5/EEA-1 positive vesicles as involved in BACE1-mediated cleavage of APP (Rajendran et al., 2006). Now, this work connects another effector of Rab5 with APP processing and provides novel insight into how APP processing could be coupled with enlargement of early endosomes. In unpublished work from my lab, we have evidence that silencing of APPL1 reduces Ab levels, thereby providing additional support to the Nixon lab findings on APPL and AD. Overall, an excellent paper on the cell biology and biochemistry of AD.
References:
Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006 Jul 25;103(30):11172-7. PubMed.
Case Western Reserve University
There are a number of observations in AD research that are controversial or conflicting. However, one consistent observation agreed upon by all is that increased β-secretase processing of APP leads to increased risk for developing AD (brought about by multiple mechanisms: increased BACE activity during aging, mutations that make APP a better BACE substrate, increased APP gene copy number, etc.). Similarly, the “protective” mutation in APP (Jonsson et al., 2012) seems to make APP resistant to BACE cleavage (Maloney et al., 2014). This knowledge has been interpreted almost exclusively through the amyloid lens, since increased BACE processing also leads to increased amyloid-β (Aβ) levels.
However, there is growing evidence that that not all deleterious effects of increased BACE processing of APP are mediated by Aβ, and this study further confirms that notion. In this study, the group from Nathan Klein Institute presents strong data that increased levels of APP β-CTF, and not that of Aβ, cause endosomal abnormalities via APPL1 and rab5. Here they characterize this interaction at molecular level and identify APP-CTF residues involved in binding APPL1. Together with the several earlier papers from Nixon and colleagues, this study leaves no doubt that β-CTF of APP mediates deleterious effects independent of Aβ.
It is likely that increased BACE processing of APP could pose a double whammy. First, increased β-CTF levels by themselves bring about the toxic events described here. Second, γ-secretase processing of β-CTF would generate Aβ and APP Intracellular domain (AICD), which in turn causes deleterious events in vivo (tau pathology, memory impairment, neurodegeneration) independent of Aβ (Ghosal et al., 2009). Protein structure studies suggest that β-CTF is a better substrate for γ-secretase cleavage (Tian et al., 2010) and several groups have independently shown that β-CTF generates increased AICD signaling (Goodger et al., 2009; Belayaev et al., 2010; Flammang et al., 2012).
In any case, data that BACE processing of APP brings about AD-related deleterious effects independent of Aβ are becoming so compelling that one cannot ignore their implications for drug development. There is growing evidence that it may be unrealistic to expect that targeting Aβ alone will be effective against AD, even if the anti-amyloid treatment is started very early during disease progression.
References:
Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer's disease and age-related cognitive decline. Nature. 2012 Aug 2;488(7409):96-9. PubMed.
Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P, van der Brug M, Liu Y, Ernst JA, Watts RJ, Atwal JK. Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem. 2014 Nov 7;289(45):30990-1000. Epub 2014 Sep 24 PubMed.
Ghosal K, Vogt DL, Liang M, Shen Y, Lamb BT, Pimplikar SW. Alzheimer's disease-like pathological features in transgenic mice expressing the APP intracellular domain. Proc Natl Acad Sci U S A. 2009 Oct 27;106(43):18367-72. PubMed.
Tian Y, Crump CJ, Li YM. Dual role of alpha-secretase cleavage in the regulation of gamma-secretase activity for amyloid production. J Biol Chem. 2010 Oct 15;285(42):32549-56. PubMed.
Goodger ZV, Rajendran L, Trutzel A, Kohli BM, Nitsch RM, Konietzko U. Nuclear signaling by the APP intracellular domain occurs predominantly through the amyloidogenic processing pathway. J Cell Sci. 2009 Oct 15;122(Pt 20):3703-14. PubMed.
Belyaev ND, Kellett KA, Beckett C, Makova NZ, Revett TJ, Nalivaeva NN, Hooper NM, Turner AJ. The transcriptionally active amyloid precursor protein (APP) intracellular domain is preferentially produced from the 695 isoform of APP in a {beta}-secretase-dependent pathway. J Biol Chem. 2010 Dec 31;285(53):41443-54. PubMed.
Flammang B, Pardossi-Piquard R, Sevalle J, Debayle D, Dabert-Gay AS, Thévenet A, Lauritzen I, Checler F. Evidence that the Amyloid-β Protein Precursor Intracellular Domain, AICD, Derives From β-Secretase-Generated C-Terminal Fragment. J Alzheimers Dis. 2012 Jan 1;30(1):145-53. PubMed.
NIMH
Endocytic pathway abnormalities have long been implicated in excessive Aβ deposition in Alzheimer's disease (AD) and Down's syndrome (DS). In particular, both endocytosis and recycling have been shown to be persistently activated, with enlarged endosomes in the AD brain associated with increased recycling (rab4) and internalization (rab5) markers. Given the special importance of endocytosis in the processing of APP and the production of Aβ, this topic has been a matter of intense research and discussion over the years. Importantly, aberrations of membrane turnover in AD have been documented in both familial and sporadic cases, with rab5 upregulation being one of the earliest neuronal responses. Despite these important recent developments, the specific mechanisms underlying these processes and their implications for neuronal biology and neurodegeneration remain poorly defined.
In this study, Kim et al., led by Ralph Nixon, address two important questions: Firstly, why is the endosomal regulator rab5 persistently activated in AD models? And secondly, how does enhanced endocytosis impact the biology and integrity of axons? Adaptor protein APPL1 and the β C-terminal fragment of APP (βCTF) are shown to be the key culprits! Indeed, excessive βCTF appears to recruit more APPL1 to the endocytic (rab5 positive) endosomes and through this causes persistent activation of GTP-rab5. This, in turn, ramps up endocytosis, leading to the production of oversized endosomes. While the functional significance of enhanced endocytosis under elevated βCTF remains unclear, Kim et al. present convincing data for its strong pathological impact: Larger endosomes pose a major challenge to the axonal transport system, causing traffic jams, swelling, and dystrophies. Whether and to what extent these processes contribute to the pathobiology of sporadic AD remains to be established.
Make a Comment
To make a comment you must login or register.