Cell-Specific Enhancer Atlas Centers AD Risk in Microglia. Again.
Quick Links
Most GWAS hits lie in noncoding regions of the genome, making it hard to figure out what they do. To address this, researchers led by Christopher Glass at the University of California, San Diego, analyzed epigenetic data from purified populations of neurons, astrocytes, oligodendrocytes, and microglia from the young human cortex. They catalogued which regulatory regions were active in each cell type, and then looked for associations with known risk loci for neurological and psychiatric diseases. In the November 14 Science, they report that while risk variants for psychiatric diseases mostly lie in neuron-specific regulatory regions, Alzheimer’s risk variants are concentrated in microglial enhancers. In other words, many polymorphisms associated with AD affect gene expression only in microglia. The findings add to recent data suggesting that these cells drive Alzheimer’s pathogenesis.
- Scientists map enhancer-promoter interactions in four cell types of healthy brain.
- This atlas places most AD risk variants in microglial-specific enhancers.
- For example, the causal risk variant for BIN1 affects its expression only in microglia.
“This is giant,” said Oleg Butovsky at Brigham and Women’s Hospital in Boston. “They use cutting-edge technology to study human data, and open up a door to understanding how microglia initiate Alzheimer’s disease.”
In addition to these Alzheimer’s findings, the dataset could enable researchers to generate and test hypotheses for neurological disease in general, Glass noted. “This is the first atlas of genetic regulatory elements specific for the major cell types of the human brain,” he told Alzforum. Alison Goate at the Icahn School of Medicine, Mount Sinai, New York, agreed. “This provides an important resource for people working on brain disorders,” she said.
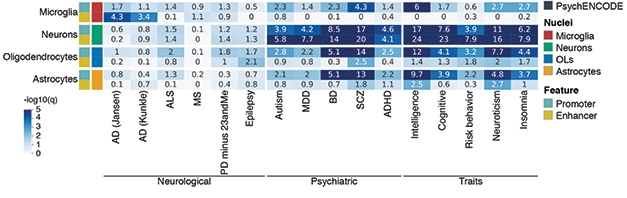
Microglia and Alzheimer’s. AD variants from two GWASs are associated almost exclusively with enhancers in microglia (left two columns, darker blue), while variants associated with psychiatric disorders and behavioral traits cluster strongly in neuronal regulatory regions (center and right column groupings). [Courtesy of Nott et al., Science/AAAS.]
Goate previously examined how AD risk variants affect gene expression, but had to use monocytes and macrophages as a proxy for microglia, because epigenetic data on these brain cells were not yet available. She found that AD risk factors were preferentially located in myeloid-specific enhancers, in agreement with the new findings (Aug 2019 news). For their part, Glass and colleagues previously isolated microglia from healthy human brain and published transcriptomic, but not epigenetic, data on these cells (Jun 2017 news).
For the current study, first authors Alexi Nott, Inge Holtman, and Nicole Coufal purified the four major brain cell types from cortical tissue samples taken from six young people aged 4–18 who were undergoing surgery to prevent future epileptic seizures. The researchers used chromatin immunoprecipitation and sequencing (ChIP-seq) to find active enhancer and promoter regions in each cell population. Active enhancers are marked by acetylation of histone H3K27, and promoters by trimethylation of H3K4 (Creyghton et al., 2010; Heintzman et al., 2007). The scientists found that all four brain-cell types shared a similar set of active promoters, but had distinct sets of active enhancers.
When transcription factor complexes assemble at active enhancers, the DNA strand loops to bind nearby promoters, initiating transcription of those genes. To find enhancer-promoter pairings in each cell type, the authors cross-linked DNA strands that were in proximity and sequenced those regions. They found 219,509 unique interactions. These clustered by cell type, defining a distinct set of enhancers that controlled the genes characteristic of each cell population.
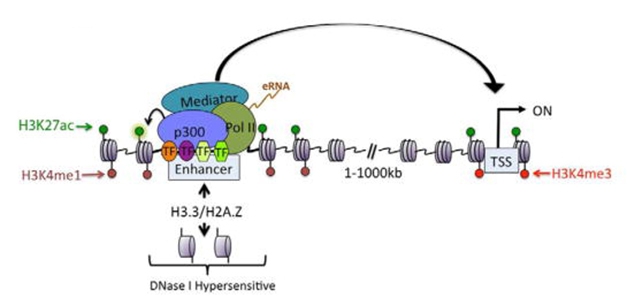
Distant Pairings Modifications to histone proteins (green and red dots) mark active enhancers, where a transcription complex (colored circles) assembles on a chromosome. DNA then loops over (arrow) to contact an active promoter (gray rectangle) and initiate transcription. [Courtesy of Calo and Wysocka, 2013.]
With this information in hand, the researchers examined disease-linked GWAS variants. Most polymorphisms associated with psychiatric disorders and behavioral traits such as autism, schizophrenia, neuroticism, and risk behavior were located in neuronal enhancers and promoters. A few of them appeared in glial promoters as well. For Alzheimer’s disease, on the other hand, GWAS risk variants were enriched only in microglial enhancers (see image above).
These microglial enhancers regulated numerous genes associated with AD, such as ABCA7, SORL1, and TREM2. The genes formed a protein interaction network centered around ApoE, the main risk factor for late-onset AD (see image below).
A detailed analysis of microglial enhancer-promoter interactions turned up some surprises. For example, AD risk variants in the SLC24A4 locus did not affect transcription of that gene, but instead mapped to promoters that controlled expression of the genes ATXN3, TRIP11, and CPSF2. This information will guide research on the functional effects of risk variants, Glass noted.

Hello Again. Alzheimer’s risk genes expressed in microglia, which have been identified in one (gray), two (green), or three (yellow) GWAS studies, and several genes with possible causal variants identified in this study (diamonds). They encode proteins that form an interaction network. [Courtesy of Nott et al., Science/AAAS.]
Although GWAS risk SNPs associate with disease, they are not necessarily the causal variant. To look for them, the authors used “fine mapping” methods pioneered by geneticists at the University of California, Los Angeles. These methods leverage the strength of disease associations to pinpoint the most likely causal variant within a locus (Kichaev et al., 2014). In 13 AD loci, the authors identified a risk variant with a high likelihood of being causal. Eight of these occurred in microglial-specific enhancers that regulated gene promoters.
One of them, the rs6733839 SNP in the BIN1 locus, had a 97 percent chance of being the causal variant. This SNP is notable for conferring a high risk of AD, second only to the ApoE4 allele among common GWAS hits. The SNP lies in a microglial-specific enhancer that binds the BIN1 promoter. The authors confirmed this microglial specificity in cultured human neurons, astrocytes, and microglia derived from induced pluripotent stem cells. Deletion of the enhancer region abolished BIN1 expression in microglia, but not in the other two cell types. The findings suggest that rs6733839 exerts its effects only through microglia. Glass presented some of these BIN1 data at a 2018 Keystone conference (Jul 2018 conference news).
Glass’ epigenetic data are broadly similar to Goate’s findings from myeloid cells. “We predicted that the AD risk would be largely microglial, and they demonstrate that in this dataset,” Goate told Alzforum. The next step will be to examine how risk and protective alleles alter microglial function, and if there are common patterns, she said.
Meanwhile, Glass is interested in how disease alters epigenetic interactions in brain cells. He noted that many AD risk alleles did not map to any enhancers or promoters in his study. One possibility is that they are expressed in other brain cell types, such as endothelial cells. It could also be that some enhancers become active in the context of disease, and all the cells in this study were isolated from brains of young people without Alzheimer’s. Glass and colleagues are now developing protocols for analyzing cells from postmortem brain samples taken from people who died of various neurodegenerative diseases. “I think that will give us a tremendous amount of information about pathological signaling pathways in the brain,” Glass said.
Butovsky believes the new data reinforce other recent findings suggesting that ApoE wreaks its havoc mostly through microglia (Sep 2017 news; Oct 2019 news). “We need to focus on microglial biology in Alzheimer’s. This is the primary target for therapy right now,” he said.—Madolyn Bowman Rogers
References
News Citations
- AD Genetic Risk Tied to Changes in Microglial Gene Expression
- What Makes a Microglia? Tales from the Transcriptome
- A Delicate Frontier: Human Microglia Focus of Attention at Keystone
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- In Tauopathy, ApoE Destroys Neurons Via Microglia
Paper Citations
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010 Dec 14;107(50):21931-6. Epub 2010 Nov 24 PubMed.
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007 Mar;39(3):311-8. Epub 2007 Feb 4 PubMed.
- Kichaev G, Yang WY, Lindstrom S, Hormozdiari F, Eskin E, Price AL, Kraft P, Pasaniuc B. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 2014 Oct;10(10):e1004722. Epub 2014 Oct 30 PubMed.
Further Reading
Papers
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why?. Mol Cell. 2013 Mar 7;49(5):825-37. PubMed.
Primary Papers
- Nott A, Holtman IR, Coufal NG, Schlachetzki JC, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, Keulen Z, Pasillas MP, O'Connor C, Nickl CK, Schafer ST, Shen Z, Rissman RA, Brewer JB, Gosselin D, Gonda DD, Levy ML, Rosenfeld MG, McVicker G, Gage FH, Ren B, Glass CK. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science. 2019 Nov 29;366(6469):1134-1139. Epub 2019 Nov 14 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
VIB-Center for Molecular Neurology
This is an extraordinary study that provides a final piece of evidence for the causal involvement of microglia in Alzheimer’s disease. This work follows up a previous report from the same group, where the authors performed transcriptomic analysis of human microglia isolated from surgical resections (Gosselin et al., 2017). Whereas that report focused on gene expression and critical differences between mouse and human transcriptomes, especially within the context of Alzheimer’s disease risk genes, the present study presents first evidence of functional impact of genetic polymorphisms in noncoding regions in microglia. Previous work by Novikova et al., already reported similar observations in monocyte/macrophage populations (Novikova et al., 2019), but the work from the Glass lab takes this a step further by studying the effect of modifications in those regulatory elements in isolated microglia.
One of the most exciting observations is perhaps the microglial-specific effect of deleting the BIN1 enhancer region in iPSC-derived cells. This adds an additional level of complexity to the system, as it suggests that the exact same genetic polymorphism in enhancer regions may alter the expression level of target genes only in particular cell types.
In light of these findings, it is now crucial to carefully study the functional impact of these genetic variants in human systems, starting from in vitro studies using iPSC-derived microglia, but also upgrading to more complex strategies using co-cultures and xenograft models that allow one to explore the response of the cells in their native brain environment (Mancuso et al., 2019; Hasselmann et al., 2019).
References:
Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JC, Sajti E, Jaeger BN, O'Connor C, Fitzpatrick C, Pasillas MP, Pena M, Adair A, Gonda DD, Levy ML, Ransohoff RM, Gage FH, Glass CK. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017 Jun 23;356(6344) Epub 2017 May 25 PubMed.
Mancuso R, Van Den Daele J, Fattorelli N, Wolfs L, Balusu S, Burton O, Liston A, Sierksma A, Fourne Y, Poovathingal S, Arranz-Mendiguren A, Sala Frigerio C, Claes C, Serneels L, Theys T, Perry VH, Verfaillie C, Fiers M, De Strooper B. Stem-cell-derived human microglia transplanted in mouse brain to study human disease. Nat Neurosci. 2019 Dec;22(12):2111-2116. Epub 2019 Oct 28 PubMed.
Hasselmann J, Coburn MA, England W, Figueroa Velez DX, Kiani Shabestari S, Tu CH, McQuade A, Kolahdouzan M, Echeverria K, Claes C, Nakayama T, Azevedo R, Coufal NG, Han CZ, Cummings BJ, Davtyan H, Glass CK, Healy LM, Gandhi SP, Spitale RC, Blurton-Jones M. Development of a Chimeric Model to Study and Manipulate Human Microglia In Vivo. Neuron. 2019 Sep 25;103(6):1016-1033.e10. Epub 2019 Jul 30 PubMed.
Takeda
The current work from the Glass lab provides striking evidences that a significant number of SNPs associated with LOAD likely affect gene expression exclusively in microglia. In particular, in the context of BIN1, the present work confirms what we previously hypothesized, i.e., that SNP rs6733839 in the BIN1 locus would affect BIN1 expression specifically in microglia (Crotti et al., 2019).
Following up on such hypothesis, and considering the correlation between BIN1 SNPs and tau-PET levels, we investigated the role of BIN1 in microglia in the context of tau pathology. We observed that BIN1-associated, tau-containing extracellular vesicles purified from CSF of AD-affected individuals are seeding-competent. Furthermore, we showed that genetic deletion of Bin1 from microglia resulted in reduction of tau secretion via extracellular vesicles in vitro, and in decrease of tau spreading in vivo. Our observations suggest that BIN1 could contribute to the progression of AD-related tau pathology by altering tau clearance and promoting release of tau-enriched extracellular vesicles by microglia (Crotti et al., 2019).
References:
Crotti A, Sait HR, McAvoy KM, Estrada K, Ergun A, Szak S, Marsh G, Jandreski L, Peterson M, Reynolds TL, Dalkilic-Liddle I, Cameron A, Cahir-McFarland E, Ransohoff RM. BIN1 favors the spreading of Tau via extracellular vesicles. Sci Rep. 2019 Jul 1;9(1):9477. PubMed.
Cardiff University
Nott, Holtman, Coufal, and colleagues have produced a tremendous resource by applying several chromatin assays to purified human brain cell types. This builds on their previous work, which was the first to investigate the gene regulatory landscape of human ex vivo microglia (Gosselin et al., 2017). In their recent study they applied nuclear sorting techniques to purify the major cell types of the human brain. They then used several genome-wide assays of chromatin biology to map noncoding elements active in each cell type. Crucially, they identify the long-range interactions between enhancers and genes using assays of chromatin conformation. Linking distal gene regulatory elements to their targets genes is not a trivial task, and when combined with data from common variant studies of disease (e.g., GWAS) can provide crucial links between risk alleles and effector genes (e.g. Miguel-Escalada et al., 2019).
While the authors use their data to annotate a variety of neuropsychiatric disease genetic hits, they focus on Alzheimer’s disease. Their analysis shows that Alzheimer’s disease risk variants are likely to operate in microglia, consistent with previous investigations (Tansey et al., 2018). Similar to a recent study using chromatin conformation data from peripheral myeloid cells (Novikova et al., 2019), they are able to link Alzheimer’s disease risk variants to target genes. Importantly, these highlighted genes are often not those most proximal to the associated variants. Together, they nominate cell types (microglia) and target genes important for the genetic risk mechanisms of Alzheimer’s disease.
Following this large-scale analysis, they functionally validate the interaction between a risk variant containing microglial enhancer and the BIN1 promoter using genome engineering of human stem cell models. The BIN1 locus contains one of the most significantly associated common risk variants for Alzheimer’s disease, but formal links to BIN1 have been lacking. As the authors note, BIN1 is expressed in multiple cell types. However, the risk mechanism appears to be specific to microglia. This information is critical for appropriate downstream biological investigation. Those interested in the biology of Alzheimer’s disease risk genes will need to consider their nominated genes and model systems carefully.
This study offers much-needed progress along the difficult path from statistical association to biological investigation. However, many risk loci are still poorly annotated. Undoubtedly, similar data from additional cell types and states will be required to fully resolve the genetic risk mechanisms of Alzheimer’s disease. Nevertheless, these data provide high-quality microglial gene targets relevant to the pathogenesis of Alzheimer’s disease that will accelerate biological investigations.
References:
Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JC, Sajti E, Jaeger BN, O'Connor C, Fitzpatrick C, Pasillas MP, Pena M, Adair A, Gonda DD, Levy ML, Ransohoff RM, Gage FH, Glass CK. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017 Jun 23;356(6344) Epub 2017 May 25 PubMed.
Novikova G, Kapoor M, TCW J, Abud EM, Efthymiou AG, Cheng H, Fullard JF, Bendl J, Roussos P, Poon WW, Hao K, Marcora E, Goate AM. Integration of Alzheimer’s disease genetics and myeloid cell genomics identifies novel causal variants, regulatory elements, genes and pathways. 2019 Jul 6. bioRxiv. BioRxiv.
Tansey KE, Cameron D, Hill MJ. Genetic risk for Alzheimer's disease is concentrated in specific macrophage and microglial transcriptional networks. Genome Med. 2018 Feb 26;10(1):14. PubMed.
Miguel-Escalada I, Bonàs-Guarch S, Cebola I, Ponsa-Cobas J, Mendieta-Esteban J, Atla G, Javierre BM, Rolando DM, Farabella I, Morgan CC, García-Hurtado J, Beucher A, Morán I, Pasquali L, Ramos-Rodríguez M, Appel EV, Linneberg A, Gjesing AP, Witte DR, Pedersen O, Grarup N, Ravassard P, Torrents D, Mercader JM, Piemonti L, Berney T, de Koning EJ, Kerr-Conte J, Pattou F, Fedko IO, Groop L, Prokopenko I, Hansen T, Marti-Renom MA, Fraser P, Ferrer J. Human pancreatic islet three-dimensional chromatin architecture provides insights into the genetics of type 2 diabetes. Nat Genet. 2019 Jul;51(7):1137-1148. Epub 2019 Jun 28 PubMed.
Institute Pasteur de Lille, INSERM
The Nott et al. paper is of particular interest for those of us who try to decipher how genes can be potentially regulated in different brain cell types through common and specific enhancers.
In particular, the authors beautifully demonstrated that an enhancer sequence is able to specifically drive BIN1 expression in microglia. This sequence of 360 pb contains the sentinel SNP (rs6733839) of the BIN1 locus, and it is tempting to consider that it is therefore the causal variant because of the convergence of these genetic and biological data. However it is important to note that the real functionality of this SNP has not been assessed in this paper. For this purpose, it would have been necessary to develop iPSC-derived microglia, neurons, and astrocytes specifically mutated for this variant, and to compare the BIN1 expression level in the mutated and corresponding isogenic cells.
There is no data showing that this variant is able to modulate the microglia enhancer activity. It is thus an overstatement to claim that rs6733839 is the causal variant.
In addition, we previously reported that the BIN1 causal variant is likely located within a 6.7 kb region encompassing rs6733839 (Chapuis et al., 2013). In this region, we detected three SNPs reaching genome-wide significant level, including rs6733839. However, only one of them, rs59335482, an insertion-deletion variant, was able to modulate the luciferase activity assay in different cell types, including a neuroblastoma cell line (keeping in mind all the limitations of such an approach). This variant unfortunately is not imputed in the IGAP database and Nott et al. have likely missed this information. However, this may, for instance, explain in part why Grubman et al. found no change in microglial BIN1 expression in the entorhinal region of AD cases when compared with controls.
References:
Chapuis J, Hansmannel F, Gistelinck M, Mounier A, Van Cauwenberghe C, Kolen KV, Geller F, Sottejeau Y, Harold D, Dourlen P, Grenier-Boley B, Kamatani Y, Delepine B, Demiautte F, Zelenika D, Zommer N, Hamdane M, Bellenguez C, Dartigues JF, Hauw JJ, Letronne F, Ayral AM, Sleegers K, Schellens A, Broeck LV, Engelborghs S, De Deyn PP, Vandenberghe R, O'Donovan M, Owen M, Epelbaum J, Mercken M, Karran E, Bantscheff M, Drewes G, Joberty G, Campion D, Octave JN, Berr C, Lathrop M, Callaerts P, Mann D, Williams J, Buée L, Dewachter I, Van Broeckhoven C, Amouyel P, Moechars D, Dermaut B, Lambert JC, GERAD consortium. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry. 2013 Nov;18(11):1225-34. Epub 2013 Feb 12 PubMed.
Roqué PJ, Dao K, Costa LG. Microglia mediate diesel exhaust particle-induced cerebellar neuronal toxicity through neuroinflammatory mechanisms. Neurotoxicology. 2016 Sep;56:204-214. Epub 2016 Aug 16 PubMed.
Make a Comment
To make a comment you must login or register.