Behold PANTHOS, a Toxic Wreath of Perinuclear Aβ That Kills Neurons
Quick Links
In Alzheimer’s disease, defective autophagy seeds Aβ fibrils around the nuclei of neurons, which then burst, leaving amyloid plaques in their wake. This is according to researchers led by Ralph Nixon and Ju-Hyun Lee of New York University, Orangeburg. In the June 2 Nature Neuroscience, they reported that in five mouse models of amyloidosis, poorly acidified lysosomes stuffed with Aβ distort the plasma membranes of neurons and form toxic rosettes around their nuclei. Dubbed PANTHOS, these flower-shaped blebs seed Aβ fibrils and leak proteases into the cytoplasm. The neurons eventually burst, leaving behind amyloid cores that tally with plaque load. PANTHOS, aka poisonous anthos, from the Greek for flower, occurs in AD brain tissue as well.
- Aβ accumulates in lysosomes when they fail to acidify.
- As the vesicles swell, they hijack Golgi and ER membranes.
- Perinuclear rosettes of Aβ fibrils then form.
- When neurons burst, the rosettes coalesce into amyloid plaques.
“This meticulously conducted work is a tour de force,” said George Perry, University of Texas, San Antonio. “It changes the paradigm by clearly showing that amyloid accumulation occurs within neurons and that aggregates come out after the neurons degenerate or die.”
Charles Glabe, University of California, Irvine, agreed. “This inside-out view has largely been ignored for at least 30 years, while the outside-in paradigm has dominated,” he told Alzforum. Glabe and others had previously proposed that intracellular Aβ kills neurons and creates plaques in the brains of people with mild cognitive impairment, though how this happened was a mystery (Mar 2013 conference news; Gouras et al., 2000; D’Andrea et al., 2001). “The issue of whether Aβ pathology starts from the inside-out, or outside-in, is one of the most important questions that needed to be resolved,” he said.
Nixon and colleagues discovered PANTHOS when studying the role of the autophagy/lysosomal system in Alzheimer’s. Scientists have become increasingly interested in the role of this degradation pathway in AD and other neurodegenerative diseases, but have been stymied tracking changes in autophagic lysosome in the brain. To overcome this, first author Lee created transgenic animals expressing the autophagy marker LC3 coupled with two dyes: one that fluoresces red when lysosomes are acidic, and the other that emits green light only when the pH creeps above 6.0. Lysosomes that fail to acidify appear yellow. A Thy-1 promoter ensured that only neurons made this tandem fluorescent LC3 reporter (tfLC3).
Lee crossed these tfLC3 mice with five models of amyloidosis: TgCRND8, PS/APP, 5xFAD, Tg2576, and APP51 (Herzig et al., 2004). The first three begin to develop amyloid plaques by about 5 months, but for Tg2576 and APP51 they form at about 12 and 20 months, respectively.
Using confocal microscopy, the scientists measured fluorescence within cortical slices from the transgenic mice months before they developed plaques. Compared to tfLC3 controls, transgenics had four times as many poorly acidified autolysosomes. “The most striking finding was that autolysosomal pH in neurons was increased long before the accumulation of amyloid plaques,” Tim Sargeant, South Australian Health and Medical Research Institute, Adelaide, wrote.
The authors thought a faulty vATPase was to blame. These pump protons into lysosomes, and their activity fell 35 percent before plaques first appeared and by half by the time they had become abundant.
When the lysosome pH stays high, proteases cannot clear cellular debris, such as Aβ. Indeed, 40 percent of cortical neurons from young, pre-plaque mice contained puncta of β-CTF/Aβ. Almost all were within the poorly acidified autolysosomes. Lee told Alzforum that the vesicles also included short, partially formed fibrils of Aβ.
As the mice aged, the autophagic process backed up. Swollen vesicles around the nucleus bulged the plasma membrane, morphing the cell body into a PANTHOS shape with distended lysosome petals around the nucleus (image below). These neurons appeared in all five mouse models. Zhenyu Yue, Icahn School of Medicine at Mount Sinai, New York, was surprised by the extreme extent that autolysosomes packed into the PANTHOS petals. “It suggests massive upregulation and impairment of autophagy,” he told Alzforum.
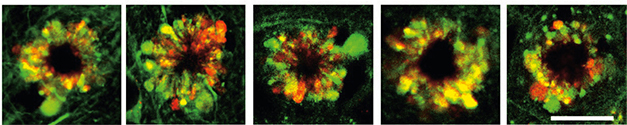
Deadly Flower. The PANTHOS structure, comprising a nuclear center (black) surrounded by blebs of poorly acidified autolysosomes (yellow) forms in Tg2576 (left), 5xFAD (second), TgCRND8 (middle), PS/APP (fourth), and APP51 (right) mice. [Courtesy of Lee et al., Nature Neuroscience, 2022.]
To get a better look at this phenomenon, the scientists homed in on cortical neurons from 5xFAD/LC3 mice. In 2.5-month-old animals that were just developing plaques, Aβ fibrils accrued within the failing autolysosomes and in tubules surrounding the nucleus. Lee said immunostaining showed that these tubules formed from the endoplasmic reticulum membrane. Autophagic vesicles can syphon from the ER to support their own swelling membranes (Uemura et al., 2014; Hayashi-Nishino et al., 2010; Axe et al., 2008). The authors reasoned that this happens in the AD mice. Alas, the autophagosomes floundered, leaving the tubules to linger and fill with Aβ fibrils.
Why has nobody detected these perinuclear Aβ fibrils before? Nixon said that without three-dimensional reconstruction of the neuron, it would be extremely difficult to tell if the fibrils lay within or outside the cell. “Only after correlating fluorescence and electron microscopy images of the same PANTHOS cell did we realize that the tubular structures are within an intact cell,” he told Alzforum (see three-dimensional reconstruction in movie below).
Flower Power. In mouse brain tissue, correlative light-electron microscopy, which images a sample with light and an electron beam simultaneously, renders a three-dimensional image of one PANTHOS neuron with a nuclear center (false-colored blue) surrounded by swollen autolysosome petals (red). The rest of the neuron is colored yellow. [Courtesy of Lee et al., Nature Neuroscience, 2022.]
As the PANTHOS neurons deteriorated, nearly all contained a dense amyloid core. In 2.5-month-old mice, half of the PANTHOS neurons were thioflavin S-positive, while by 6 months almost all were, by which time the animals had full-blown plaque pathology. “PANTHOS neurons look like plaques, even though they’re intact cells,” Nixon said. Remarkably, the scientists noticed that some Thio-S-positive PANTHOS neurons had burst, and their contents seemed to mingle with debris from neighboring cells to create a large amyloid plaque with multiple dense-cores (image below).

From PANTHOS to Plaques. In a brain tissue slice from a mouse, three PANTHOS neurons filled with autolysosomes (red) and each carrying a Thio-S-positive core (blue) burst and coalesced to form one large amyloid plaque with multiple dense cores. [Courtesy of Lee et al., Nature Neuroscience, 2022.]
Perry and Gunnar Gouras, Lund University, Sweden, emphasized that, while some plaques may form from neuronal soma this way, others arise from dystrophic neurites and synapses (reviewed by Gouras et al., 2013). “Synapses are affected early in AD, and we have seen synaptic endosomes as the earliest sites of APP β-CTF/Aβ damage in AD that leads to early synaptic dysfunction, massive autophagic vacuole accumulation, and neuron death,” he wrote (full comment below). Still, Lee found that in mice, at least, the majority of plaques formed via PANTHOS. Using the 3D6 antibody to label plaques, Lee found that all the antibody binding was accounted for in PANTHOS lesions.
Do plaques form through PANTHOS in people, too? Analysis of prefrontal cortex tissue from three adults who had died at Braak stage II revealed the same perinuclear autophagic lysosomes and amyloid fibrils in some neurons, though the authors did not quantify how many. Nixon said that work is ongoing.
In all, these findings suggest that, months before plaques develop in mice, intraneuronal Aβ accumulates in faulty lysosomes, and that this distorts and kills neurons, leaving behind amyloid plaques (image below). “This study provides strong support that lysosome dysfunction is an early, causal, and, most importantly, pathogenic process in Alzheimer’s disease,” Rick Livesey, University College London, wrote (full comment below)..

PANTHOS Progression. This model shows how poorly acidified autolysosomes (purple) gather in neuron somas (left), pushing out the cell membrane to form PANTHOS petals (middle). Lysosomes begin leaking proteases (pink) and Aβ fibrils (gray squiggles) aggregate in lysosomal tubules around the nucleus. The overstuffed neuron collides with nearby PANTHOS cells and ruptures (right), recruiting glia to turn the amyloid debris into larger plaques. [Courtesy of Lee et al., Nature Neuroscience, 2022.]
In older mice, microglia swarmed PANTHOS neurons that had burst, and the authors think that these glia help to mop up diffuse Aβ and help package it into the dense core plaques, as has been reported previously (Apr 2021 news; May 2016 news). They also believe that correcting the lysosomal pH deficit might prevent these downstream pathologies. “It would make more sense for therapeutics to target the autophagy-lysosome pathway (the cause) rather than solely amyloid plaques (the effect), as is the case with current clinical trials that use Aβ immunotherapy by itself,” Sargeant wrote. Glabe agreed. “If the plaque is the tombstone marker for neuritic neurons, then removing it won’t raise the dead,” he said.
In preliminary experiments, Lee is trying to rescue lysosomal function by giving 5xFAD mice isoproterenol. He recently found that this β-adrenergic agonist, used to treat heart rhythm problems, promotes lysosome acidification and function in AD patient-derived fibroblasts (Lee et al., 2020). So far, he has found that animals treated with isoproterenol have fewer PANTHOS neurons.—Chelsea Weidman Burke
References
News Citations
- Like Star Born of Supernova, Plaque Born of Exploded Neuron?
- Microglia Build Plaques to Protect the Brain
- Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
Research Models Citations
Paper Citations
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000 Jan;156(1):15-20. PubMed.
- D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001 Feb;38(2):120-34. PubMed.
- Herzig MC, Winkler DT, Burgermeister P, Pfeifer M, Kohler E, Schmidt SD, Danner S, Abramowski D, Stürchler-Pierrat C, Bürki K, van Duinen SG, Maat-Schieman ML, Staufenbiel M, Mathews PM, Jucker M. Abeta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat Neurosci. 2004 Sep;7(9):954-60. PubMed.
- Uemura T, Yamamoto M, Kametaka A, Sou YS, Yabashi A, Yamada A, Annoh H, Kametaka S, Komatsu M, Waguri S. A cluster of thin tubular structures mediates transformation of the endoplasmic reticulum to autophagic isolation membrane. Mol Cell Biol. 2014 May;34(9):1695-706. Epub 2014 Mar 3 PubMed.
- Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. Electron tomography reveals the endoplasmic reticulum as a membrane source for autophagosome formation. Autophagy. 2010 Feb;6(2):301-3. Epub 2010 Feb 6 PubMed.
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008 Aug 25;182(4):685-701. PubMed.
- Gouras GK, Willén K, Faideau M. The Inside-Out Amyloid Hypothesis and Synapse Pathology in Alzheimer's Disease. Neurodegener Dis. 2013 Sep 24; PubMed.
- Lee JH, Wolfe DM, Darji S, McBrayer MK, Colacurcio DJ, Kumar A, Stavrides P, Mohan PS, Nixon RA. β2-adrenergic Agonists Rescue Lysosome Acidification and Function in PSEN1 Deficiency by Reversing Defective ER-to-lysosome Delivery of ClC-7. J Mol Biol. 2020 Apr 3;432(8):2633-2650. Epub 2020 Feb 24 PubMed.
Further Reading
Papers
- Cataldo AM, Hamilton DJ, Nixon RA. Lysosomal abnormalities in degenerating neurons link neuronal compromise to senile plaque development in Alzheimer disease. Brain Res. 1994 Mar 21;640(1-2):68-80. PubMed.
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D. Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease. J Neurosci. 1996 Sep 15;16(18):5795-811. PubMed.
- Im E, Jiang Y, Stavrides P, Darji S, Erdjument-Bromage H, Neubert TA, Bordi M, Bordi M, Choi JY, Lee JH, Nixon RA, Choi JY, Lee JH, Nixon RA. Lysosomal dysfunction in Down Syndrome and Alzheimer mouse models is caused by selective v-ATPase inhibition by Tyr682 phosphorylated APP βCTF. bioRxiv, June 4, 2022 bioRxiv
News
- Lodged in Late Endosomes, Presenilin 2 Churns Out Intraneuronal Aβ
- Are Intraneuronal Aβ Oligomers ‘Seeding Units’ of Alzheimer’s Disease?
- Mistakes Prompt Retraction of Controversial Paper, and Publication Ban
- Partners in Crime: APP Fragment and Endosomal Protein Impair Endocytosis
- Aβ Oligomers Linked to ER Stress in Patient-Derived Neurons
- Wiping Out Microglia Prevents Neuritic Plaques
Primary Papers
- Lee JH, Yang DS, Goulbourne CN, Im E, Stavrides P, Pensalfini A, Chan H, Bouchet-Marquis C, Bleiwas C, Berg MJ, Huo C, Peddy J, Pawlik M, Levy E, Rao M, Staufenbiel M, Nixon RA. Faulty autolysosome acidification in Alzheimer's disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat Neurosci. 2022 Jun;25(6):688-701. Epub 2022 Jun 2 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Adelaide
The relationship between amyloid plaques and associated abundant lysosomal protein staining has been widely observed for decades (Whyte et al., 2020). However, the details behind the association remained vague. This was frustrating because plaques, and markers of extreme lysosomal dysfunction, co-localized in space and time and were so obviously causally linked. This study from Lee and colleagues represents a conceptual leap forward in this area, and it points the way for the development of therapies.
One important part of this study is that it shows that plaque-associated lysosomal system cargo comes from enlarged blebs that largely originate from the neuronal perikaryon, and not predominantly from dystrophic (swollen) neurites. Further, the formation of these neuronal perikaryon-derived blebs, named “PANTHOS,” gives rise to amyloid plaques themselves. Although this observation is important, it is not the most important part of the study by Lee and colleagues.
The most striking finding was that autolysosomal pH in neurons increased long before the accumulation of amyloid plaques. This has important implications. In Alzheimer’s disease, it appears as that amyloid plaques are the sequelae of autophagy-lysosomal pathway impairment. Research from rare diseases tells us that lysosomal system dysfunction by itself is capable of initiating progressive neurodegeneration—mutations that impair the initiation of autophagy, or lysosomal hydrolysis, are both capable of creating neurodegenerative conditions in humans (Collier et al., 2021; Cachon-Gonzalez et al., 2018). It would thus make more sense for therapeutics to target the autophagy-lysosome pathway (the cause) rather than solely amyloid plaques (the effect), as is the case with current clinical trials that utilize Aβ immunotherapy alone.
When would lysosome-targeting therapies work the best? We know that amyloid plaques (the outcome of lysosomal system impairment) can begin to form in people two decades before a diagnosis of dementia (Roberts et al., 2017) and Lee and colleagues show that lysosomal system dysfunction is present long before the deposition of amyloid plaques in mice. So how long before a dementia diagnosis do lysosomes in a person’s brain begin to struggle to maintain a low pH? Further, how do well-known mid-life environmental risk factors for dementia, such as obesity, impact lysosomal acidification? Answers to these questions will get us closer to interventions that can delay the onset of dementia.
References:
Cachon-Gonzalez MB, Zaccariotto E, Cox TM. Genetics and Therapies for GM2 Gangliosidosis. Curr Gene Ther. 2018;18(2):68-89. PubMed.
Collier JJ, Guissart C, Oláhová M, Sasorith S, Piron-Prunier F, Suomi F, Zhang D, Martinez-Lopez N, Leboucq N, Bahr A, Azzarello-Burri S, Reich S, Schöls L, Polvikoski TM, Meyer P, Larrieu L, Schaefer AM, Alsaif HS, Alyamani S, Zuchner S, Barbosa IA, Deshpande C, Pyle A, Rauch A, Synofzik M, Alkuraya FS, Rivier F, Ryten M, McFarland R, Delahodde A, McWilliams TG, Koenig M, Taylor RW. Developmental Consequences of Defective ATG7-Mediated Autophagy in Humans. N Engl J Med. 2021 Jun 24;384(25):2406-2417. PubMed.
Roberts BR, Lind M, Wagen AZ, Rembach A, Frugier T, Li QX, Ryan TM, McLean CA, Doecke JD, Rowe CC, Villemagne VL, Masters CL. Biochemically-defined pools of amyloid-β in sporadic Alzheimer's disease: correlation with amyloid PET. Brain. 2017 May 1;140(5):1486-1498. PubMed.
Whyte LS, Hassiotis S, Hattersley KJ, Hemsley KM, Hopwood JJ, Lau AA, Sargeant TJ. Lysosomal Dysregulation in the Murine AppNL-G-F/NL-G-F Model of Alzheimer's Disease. Neuroscience. 2020 Mar 1;429:143-155. Epub 2020 Jan 7 PubMed.
Lund University
This is wonderful work by Lee et al. using a transgenic pH-sensitive LC3 fluorescent probe to convincingly show plaque formation from within neuron cell bodies in five different AD mouse models. The extensive work that the group of Nixon and colleagues have accomplished over the years on the role of the endosome-lysosome-autophagy system in AD is extraordinary. As a longtime proponent of the importance of intraneuronal Aβ in AD, I also hope this study can reboot more AD researchers from their belief that plaques arise from the aggregation of secreted extracellular Aβ.
I had also described plaques seemingly forming from neurons in AD brains (Gouras et al., 2000), and this was shown really nicely by Mike D’Andrea and colleagues (D’Andrea et al., 2001). However, already in this first study, I was puzzled that AD-vulnerable neurons, which show the most prominent early Aβ42 accumulation in layer 2 of the entorhinal cortex, do not at all develop into plaques but rather subsequently form tangles, while the earliest hippocampal plaques develop at the axon terminals of these projection neurons. This led to our immuno-EM study, which not only showed normal localization of Aβ42 in late endosomal multivesicular bodies (MVBs) but that pre-plaque Aβ accumulation is most prominent in MVBs in distal neurites near synapses (Takahashi et al., 2002), which precedes the massive increase in autophagic vacuoles (AVs). More recently, we provided further evidence for such Aβ accumulation and plaque formation from within perforant path axonal dystrophies in the outer molecular layer of the dentate (Roos et al., 2021).
Interestingly, from over a century ago, Oskar Fischer’s beautiful drawings of plaques continue to provide clues (Fischer, 1907). He also depicted structures (“drusige Wucherungen”) like the cellular blebs (“PANTHOS”) visualized so nicely in the current Lee et al. study. However, Fischer’s drawings also highlight dystrophies and remarkably, some of the cell body blebs connect to neurites.
Neuron soma, of course, also have synaptic inputs, and increasing evidence over the years has indicated that synapses are affected early in AD. Thus, we view the synaptic endosome as the earliest sites of APP βCTF/Aβ damage in AD, which leads to early synapse dysfunction, massive AV accumulation and then neuron death.
In addition to their new insights into the origin of neuron-derived plaques with these unusual AV-filled blebs, Lee et al. provide new findings on AV de-acidification and a decline in vATPases’ activity that are of considerable interest.
Many more questions remain, such as by what mechanism(s) AV deacidification initiates, what explains its anatomic selectivity, etc. Evidence that Aβ accumulation induces endosome membrane permeability has been described, which could be a cause of the de-acidification. However, endosome-lysosome-autophagy, and other age-related dysfunctions, could also be upstream leading to APP-βCTF/Aβ accumulation.
References:
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000 Jan;156(1):15-20. PubMed.
D'Andrea MR, Nagele RG, Wang HY, Peterson PA, Lee DH. Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology. 2001 Feb;38(2):120-34. PubMed.
Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002 Nov;161(5):1869-79. PubMed.
Roos TT, Garcia MG, Martinsson I, Mabrouk R, Israelsson B, Deierborg T, Kobro-Flatmoen A, Tanila H, Gouras GK. Neuronal spreading and plaque induction of intracellular Aβ and its disruption of Aβ homeostasis. Acta Neuropathol. 2021 Oct;142(4):669-687. Epub 2021 Jul 16 PubMed.
Fischer O. Miliaere Nekrosen mit drusigen Wucherungen der Neurofibrillen, eine regelmassige Veraenderung der Hirnrinde bei seniler Demenz. Monatsschr Psychiat Neurol. 1907 Jan 1;22:361-72.
University College London
There is increasing understanding of the role of endolysosomal and autophagy dysfunction in neurodegenerative diseases, including Alzheimer’s disease, and bona-fide lysosomal storage disorders, such as Niemann-Pick disease. This is a tour de force study from the Nixon group on lysosome and autophagy function in five different mouse models for monogenic AD. It provides strong support to the school of thought that lysosome dysfunction is an early, causal, and, most importantly, pathogenic process in Alzheimer’s disease, not a secondary consequence of protein aggregation.
The authors’ detailed and thorough analyses of the different models also indicate that Aβ aggregation and amyloid plaque formation begin intracellularly in vivo, providing very useful insights to help resolve a long-running debate about the genesis of amyloid plaques.
A striking finding of the paper is that reduced lysosome acidification is an early event in neurons in the mouse models, long before the appearance of any amyloid plaques or aggregates, and that it is accompanied by accumulation of the C-terminus of APP (the product of β-secretase proteolysis of APP, referred to as β-CTF) and Aβ peptides. Previous studies in cellular models from this group, and in human iPSC-derived neurons by others in the field, have shown that altered APP processing and accumulation of β-CTFs are causal for lysosome dysfunction, with subsequently reduced autophagy.
The present study makes creative use of mouse models and high-content imaging to map out the temporal evolution of lysosome and autophagy function, alongside the accumulation of Aβ fibrils and morphological changes in neurons. Together, the data contribute compelling evidence to the hypothesis that plaque formation is a later pathological consequence of earlier lysosome and autophagy dysfunction, at least in monogenic AD.
Furthermore, this study also suggests that the mouse models might be revisited as informative models for the early stages of AD, before plaque formation, both for mechanistic studies and for investigating candidate disease-modifying or preventative treatments.
Columbia University
This is a remarkably important study, which, if true in AD, will have an enormous impact on the field.
To date, nearly all hypotheses on plaques have been based on the idea that there are changes in secretion of the peptide from neurons under stress. Here, Nixon et al. provide very strong evidence that plaques are instead remnants of dying neurons, and that this occurs with damage to normal autophagic and lysosomal function.
Further, they have some evidence, including from their earlier papers, that at some level, this is due to a loss of the acidified environment required for activation of degradative enzymes in lysosomes.
The cause of this lack of normal autolysosomal function is not yet clear. But if true—and the evidence looks solid—it would be a discovery on par with that in the field of ulcer biology, when that biology was discovered to be due to bacterial infection, and it will change most basic and clinical approaches to AD.
Johannes Gutenberg-University Mainz, Medical School
By establishing and combining elegant tools, such as the transgenic dual-fluorescence probe that allows the identification of autophagy compartments and pH changes in vivo, with an extensive range of histochemistry and imaging techniques, Lee et al. establish in five frequently used APP-AD mouse models that the early autophagy failure is initiated by pathologically decreased levels of autophagosome/lysosome acidification. Interestingly, autolysosome acidification declines well before the deposition of extracellular amyloid.
Further, more compromised yet intact neurons display a striking autophagic stress response, characterized by massive poorly acidified autophagic vacuoles that peripheralize and protrude the plasma membrane to form Aβ/APP-bCTF-filled flower-like blebs, thus named PANTHOS. Strikingly, PANTHOS is not only identified in the mouse models but also in preliminary studies in human AD brain tissue (neocortical neurons), further highlighting the significance of the findings.
Most importantly, this in-vivo study demonstrates that PANTHOS exhibited by individual neurons, which first generates an intraneuronal Aβ core, is, in fact, the principal origin of the majority of amyloid-containing senile plaques in the AD mouse models studied. These data suggest that Aβ deposition and plaque formation are the consequence of an already highly neurodegenerative pathology rather than the cause, supporting a sequence of pathological events that is fundamentally different from the widely acknowledged amyloid-cascade hypothesis that has been around for 30 years.
Further support for the pathogenic relevance of autophagic disturbances in humans comes from the fact that deterioration of autophagy, and autophagic stress, is a hallmark of many congenital lysosomal storage disorders that also show other pathological overlaps with AD. Selective macroautophagy pathways are of key importance for maintaining neuronal survival and function; a targeted modulation may increase the resilience of neurons, especially during aging.
Taken together, these new and exciting findings point toward novel pharmacological targets, such as ameliorating the pH deficit, (which is already being investigated by the authors and shows alleviating effects, rather than the quantitative removal of amyloid as currently favored.
Early endosomal-lysosomal changes/disturbances as a leading cause in the pathogenesis of AD have been hypothesized and demonstrated time and again through many studies by the Nixon group, as well as others. It is about time for us to finally, and stringently, follow these important leads.
University of Sussex
This paper gives us clear information regarding the source of amyloid plaques. It provides strong evidence for the generation of amyloid plaques from within dysfunctional autolysosomes via a stage named “PATHOS,” whereby large aggregates form within membrane-bound structures around the nucleus and eventually confer the characteristic amyloid plaque appearance.
Importantly, early effects of the accumulating APP cleavage products (Aβ and CTF) are very apparent, and help to explain our general view that early intermediates are most deleterious. The authors show that the accumulation of Aβ and Aβ-CTF is associated with impairment of the function of autolysosomes, including reduced ATPase activity resulting in impaired reacidification and cathepsin leakage.
This work raises a number of important questions. Firstly, it has been previously discussed extensively that early diagnosis is imperative for effective treatment. This study again reiterates the importance of this point, clearly demonstrating in this mouse model that damage to neurons happens very early and long before the appearance of amyloid plaques.
It is interesting to consider here that Aβ accumulation with the neurons results in very significant disruption and damage that, in turn, results in neuronal death, leaving behind the “scar,” which is an amyloid plaque. In contrast, tau neurofibrillary tangles accumulate within neurons, effectively filling them, but the cells remain intact (if not healthy), begging the question of what is tau doing?
The University of Adelaide
Lee, Nixon, and colleagues have previously made significant contributions to understanding the molecular and cellular mechanisms underlying AD, such as Nixon's early demonstration of changes in autophagosomes/lysosomes in AD brains (Cataldo et al., 1996; Yu et al., 2004; Nixon et al., 2005), the revolutionary Cell paper of 2010 showing involvement of the presenilin 1 holoprotein in assembly of the vacuolar ATPase (Lee et al., 2010), and the Jiang et al. paper of 2019 demonstrating the dose-dependent role of APP's βCTF in control of lysosomal acidification (Jiang et al., 2019). The critical role of the endolysosomal pathway function in development of AD is supported by the discovery of many genetic variants with involvement in this pathway (Gao et al., 2018) including APOE4 (Prasad et al., 2018).
Here, Lee et al. connect failure of autolysosome/lysosome acidification in neurons to formation of the neuritic plaques containing Aβ that are defined as characterizing AD. They use a transgenic reporter of autophagosome/lysosome acidity to demonstrate age-dependent failure of acidification of autolysosomes in neurons of transgenic mouse models of AD. This leads to accumulation of βCTF and Aβ in the perikaryal region of neurons, progressing to large membrane blebs that subsequently become neuritic plaques after cell death. It is fascinating how dysmorphic the affected neurons can become before cellular integrity is lost with the astrocytic/microglial invasion that ultimately results in the Aβ "fossil" remains of the mature plaque. Supplementary Video 1 showing beautiful three-dimensional imaging of a neuron displaying what the authors have named the PANTHOS ("poisonous flower") morphology is a must-view.
This beautiful work may be seen by some as supporting a neuron-centric view of AD. However, we should remember that, while transgenic mouse models of AD can certainly form neuritic plaques, by other broad measures, they fail to align with the human disease (Hargis and Blalock, 2016; Foley et al., 2015). Neurons’ normal function and survival is dependent upon their glial support cells, and it is important to investigate how autophagic and endolysosomal deficits in these cells contribute to the neuronal stress that eventually results in PANTHOS and neuritic plaque formation.
References:
Cataldo AM, Hamilton DJ, Barnett JL, Paskevich PA, Nixon RA. Properties of the endosomal-lysosomal system in the human central nervous system: disturbances mark most neurons in populations at risk to degenerate in Alzheimer's disease. J Neurosci. 1996 Jan;16(1):186-99. PubMed.
Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchiyama Y, Lamb BT, Cuervo AM, Nixon RA. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer's disease. Int J Biochem Cell Biol. 2004 Dec;36(12):2531-40. PubMed.
Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005 Feb;64(2):113-22. PubMed.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010 Jun 25;141(7):1146-58. PubMed.
Jiang Y, Sato Y, Im E, Berg M, Bordi M, Darji S, Kumar A, Mohan PS, Bandyopadhyay U, Diaz A, Cuervo AM, Nixon RA. Lysosomal Dysfunction in Down Syndrome Is APP-Dependent and Mediated by APP-βCTF (C99). J Neurosci. 2019 Jul 3;39(27):5255-5268. Epub 2019 May 1 PubMed.
Gao S, Casey AE, Sargeant TJ, Mäkinen VP. Genetic variation within endolysosomal system is associated with late-onset Alzheimer's disease. Brain. 2018 Sep 1;141(9):2711-2720. PubMed.
Prasad H, Rao R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc Natl Acad Sci U S A. 2018 Jul 10;115(28):E6640-E6649. Epub 2018 Jun 26 PubMed.
Hargis KE, Blalock EM. Transcriptional signatures of brain aging and Alzheimer's disease: What are our rodent models telling us?. Behav Brain Res. 2016 May 4; PubMed.
Foley AM, Ammar ZM, Lee RH, Mitchell CS. Systematic review of the relationship between amyloid-β levels and measures of transgenic mouse cognitive deficit in Alzheimer's disease. J Alzheimers Dis. 2015 Jan 1;44(3):787-95. PubMed.
Washington University School of Medicine
This exciting and provocative paper by Lee and colleagues proposes that neuronal intracellular accumulation of Aβ and lysosomal dysfunction in mouse models of AD cause a unique pattern of neurodegeneration that precedes and originates extracellular amyloid plaques.
The authors generated a transgenic mouse expressing an mRFP-eGFP-LC3 cassette in neurons, such that autophagosome (AP) conversion into autophagolysosomes (ALs) is paralleled by a fluorescence switch from yellow (eGFP + mRFP) to red (mRFP only) due to inactivation of eGFP in acidic pH. Lee et al. crossed these mice to different models of AD, and found that the formation of extracellular Aβ deposits is preceded by deficient acidification of ALs, decreased lysosomal vATPase activity, and intracellular accumulation of the cleaved carboxy-terminal fragment of APP (βCTF) and Aβ.
This early stages accumulation Aβ in poorly acidified ALs was associated with a novel and unique pattern of neural degeneration, called PANTHOS, which results in cell death and conversion into senile plaques.
The paper is provocative because it suggests that Aβ accumulation in AD is not an extracellular event, but rather initiates as an intracellular event, i.e., intraneuronal build-up of Aβ in ALs, which will subsequently cause dystrophy and death of neurons that generate extracellular amyloid plaques.
One implication of the study is that immunotherapy strategies aimed at Aβ removal may be insufficient to impact the progression of AD pathology. However, it is worth noting that in mouse models of Aβ plaques, mouse brains do not show marked neuronal loss in parallel to Aβ accumulation; moreover, the cognitive decline becomes perceptible decades after Aβ plaques appear in AD patients. Thus, the link between intracellular Aβ, extracellular Aβ, and neuronal death will require further study.
Another exciting point of the study is the morphology of PANTHOS degeneration. The authors propose that a centralized amyloid “core” is present within single intact PANTHOS neurons. Interestingly, studies of microglia in the AD field have suggested that one major function for microglia is to facilitate amyloid compaction (Yuan et al., 2016); moreover, long-term elimination of microglia in a mouse model of AD prevented the formation of Aβ plaques with a compact core (Spangenberg et al., 2019). Thus, it will be important to examine the interplay between PANTHOS and microglial response in defining the final configuration of the plaque.
Finally, while this study reiterates a fundamental role of neuronal autophagy in natural aging and neurogenerative disease, it will also prompt similar studies in other brain cells. A seminal study from Kim Green’s laboratory has already shown that LC3-associated endocytosis (LANDO) in microglia facilitates Aβ clearance, while impaired LANDO exacerbates Aβ and Tau deposition (Heckmann et al., 2019). Another important study from the Jagodic group has demonstrated that recovery in a model of neuroinflammation depends on microglial autophagy (Berglund et al., 2020). The study by Lee et al. suggests that the establishment of a microglia-specific transgenic mRFP-eGFP-LC3 mouse model may further advance our understanding the impact of microglia autophagy in AD.
—Shoutang Wang, a member of the Colonna Lab at Washington University, co-wrote this comment.
References:
Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J. TREM2 Haplodeficiency in Mice and Humans Impairs the Microglia Barrier Function Leading to Decreased Amyloid Compaction and Severe Axonal Dystrophy. Neuron. 2016 May 18;90(4):724-39. PubMed.
Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, Zhang Y, Spevak W, Lin J, Phan NY, Habets G, Rymar A, Tsang G, Walters J, Nespi M, Singh P, Broome S, Ibrahim P, Zhang C, Bollag G, West BL, Green KN. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer's disease model. Nat Commun. 2019 Aug 21;10(1):3758. PubMed.
Heckmann BL, Teubner BJ, Tummers B, Boada-Romero E, Harris L, Yang M, Guy CS, Zakharenko SS, Green DR. LC3-Associated Endocytosis Facilitates β-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer's Disease. Cell. 2019 Jul 25;178(3):536-551.e14. Epub 2019 Jun 27 PubMed. Correction.
Berglund R, Guerreiro-Cacais AO, Adzemovic MZ, Zeitelhofer M, Lund H, Ewing E, Ruhrmann S, Nutma E, Parsa R, Thessen-Hedreul M, Amor S, Harris RA, Olsson T, Jagodic M. Microglial autophagy-associated phagocytosis is essential for recovery from neuroinflammation. Sci Immunol. 2020 Oct 16;5(52) PubMed.
Katholieke Universiteit Leuven, Department of Imaging and Pathology, Laboratory of Neuropathology
Lee et al. investigated autolysosomal acidification using a novel, dual-tagged autophagy sensor in TRGL mouse brain. This sensor was built by pH-resistant mRFP together with pH-sensitive eGFP and LC3 (= tfLC3). Like LC3, the tfLC3 sensor is located in autophagosomes. Accordingly, this sensor allows the distinction between fully acidified and poorly acidified autophagosomes.
By crossing TRGL mice with different APP- or APP/PS1-overexpressing mouse lines, the authors studied the impact of APP or PS1 overexpression and/or plaque formation on acidification of autolysosomes, i.e., on the status of proper autophagy. Poorly acidified autophagosomes are in this context indicative of autophagy failure. In the resulting mouse lines, the authors found increased levels of poorly acidified autophagosomes in the different APP-TRGL and APP/PS1-TRGL transgenic mouse models. In addition, some neurons exhibited Aβ-positive vacuoles packed into large membrane blebs formed perikaryal, flowerlike rosettes that were termed PANTHOS by the authors. Here, poor acidification was observed. These PANTHOS features were also found in human AD autopsy brains. Accordingly, poor acidification of autolysosomes contributes to the neurodegenerative concert in the AD brain, probably in an APP/Aβ-driven manner.
With this work, Lee et al. pave the way for a better understanding of autophagy processes in the neurodegenerative concert of AD. The facts that (1) APP and APP/PS transgenic mice develop this type of autophagy impairment and (2) similar features were observed in the AD brain, argue in favor of an impairment of autophagy presumably triggered by Aβ/APP. To clarify whether APP overexpression or Aβ production is the key event, similar experiments with target replacement mice expressing “physiological” levels of mutant human APP may be interesting. A link to tau is not clear yet.
Earlier work on autophagy and cellular prion protein (PrPC) indicated that PrPC is a modifier of autophagy flux (Moon et al., 2016). In light of critical interactions between Aβ oligomers and PrPC (Cox et al., 2019; Gunther et al., 2019; Laurén et al., 2009) and its impact on neurodegeneration and the propagation of tau pathology (Corbett et al., 2020; Gomes et al., 2019), the question arises whether PANTHOS morphologies could be a parameter to measure target engagement of therapies aiming at interfering with Aβ-induced dysregulation of autophagy function, probably by targeting its interaction with PrPC.
That said, neurodegenerative processes in the AD brain are complex and include also activation of other degenerative pathways, for example the development of necroptosis activation (Caccamo et al., 2017) in granulovacuolar degeneration bodies, which is strongly associated with neuron loss in AD (Koper et al., 2020). Given that both autophagy and necroptosis pathway activation contribute to the neurodegenerative processes in AD, future research on these degenerative processes will be essential to understand the pathogenesis of AD and probably help to develop novel concepts for therapy.
References:
Caccamo A, Branca C, Piras IS, Ferreira E, Huentelman MJ, Liang WS, Readhead B, Dudley JT, Spangenberg EE, Green KN, Belfiore R, Winslow W, Oddo S. Necroptosis activation in Alzheimer's disease. Nat Neurosci. 2017 Sep;20(9):1236-1246. Epub 2017 Jul 24 PubMed.
Corbett GT, Wang Z, Hong W, Colom-Cadena M, Rose J, Liao M, Asfaw A, Hall TC, Ding L, DeSousa A, Frosch MP, Collinge J, Harris DA, Perkinton MS, Spires-Jones TL, Young-Pearse TL, Billinton A, Walsh DM. PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins. Acta Neuropathol. 2020 Mar;139(3):503-526. Epub 2019 Dec 18 PubMed.
Cox TO, Gunther EC, Brody AH, Chiasseu MT, Stoner A, Smith LM, Haas LT, Hammersley J, Rees G, Dosanjh B, Groves M, Gardener M, Dobson C, Vaughan T, Chessell I, Billinton A, Strittmatter SM. Anti-PrPC antibody rescues cognition and synapses in transgenic alzheimer mice. Ann Clin Transl Neurol. 2019 Mar;6(3):554-574. Epub 2019 Feb 27 PubMed.
Gomes LA, Hipp SA, Rijal Upadhaya A, Balakrishnan K, Ospitalieri S, Koper MJ, Largo-Barrientos P, Uytterhoeven V, Reichwald J, Rabe S, Vandenberghe R, von Arnim CA, Tousseyn T, Feederle R, Giudici C, Willem M, Staufenbiel M, Thal DR. Aβ-induced acceleration of Alzheimer-related τ-pathology spreading and its association with prion protein. Acta Neuropathol. 2019 Dec;138(6):913-941. Epub 2019 Aug 14 PubMed.
Gunther EC, Smith LM, Kostylev MA, Cox TO, Kaufman AC, Lee S, Folta-Stogniew E, Maynard GD, Um JW, Stagi M, Heiss JK, Stoner A, Noble GP, Takahashi H, Haas LT, Schneekloth JS, Merkel J, Teran C, Naderi ZK, Supattapone S, Strittmatter SM. Rescue of Transgenic Alzheimer's Pathophysiology by Polymeric Cellular Prion Protein Antagonists. Cell Rep. 2019 Jan 2;26(1):145-158.e8. PubMed.
Koper MJ, Van Schoor E, Ospitalieri S, Vandenberghe R, Vandenbulcke M, von Arnim CA, Tousseyn T, Balusu S, De Strooper B, Thal DR. Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer's disease. Acta Neuropathol. 2020 Mar;139(3):463-484. Epub 2019 Dec 4 PubMed.
Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009 Feb 26;457(7233):1128-32. PubMed.
Moon JH, Lee JH, Nazim UM, Lee YJ, Seol JW, Eo SK, Lee JH, Park SY. Human prion protein-induced autophagy flux governs neuron cell damage in primary neuron cells. Oncotarget. 2016 May 24;7(21):29989-30002. PubMed.
Juntendo University Graduate School of Medicine
The autophagy-lysosomal pathway (ALP) is important for the maintenance of cellular metabolism, while unneeded and old cytosolic components and organelles are delivered to the lysosome (LY) by autophagy. Delivery of such obsolete proteins and organelles to autolysosomes is performed by formation of autophagosomes (AP) that need double membranes for sequestration supplied mostly from Atg9A-positive vesicles and endoplasmic reticulum. After AP fuses with LY or transporting vesicles with lysosomal enzymes, it lowers pH by vATPase (proton pump) and becomes AL.
Until recently, many neurodegenerative diseases had been shown to be associated with defective autophagy, which may be related to each step of autophagy, including substrate sequestration, selective autophagy, lysosome fusion, and digestion in lysosomes. AP formation is strictly regulated by protein products of core autophagy-related genes, and their defects are associated to various diseases such as static encephalopathy of childhood with neurodegeneration in adulthood (SENDA).
In addition to AP formation deficits, impaired autophagosome maturation due to various mechanisms is also related to lower-motor-neuron disease, ALS, spinal and tubular muscular atrophy, and frontotemporal dementia. Moreover, defects of lysosomal enzymes induce lysosomal storage disorders including Gaucher disease, Krabbe disease, and neuronal ceroid lipofuscinoses (NCLs) due to tripeptidyl peptidase I and cathepsin D (CtsD). There are many causative genes for NCLs, and model mice have been produced. A mouse model due to the CtsD gene (CLN10) defect has demonstrated a clear-cut phenotype resembling that of the early infant type of NCLs.
APP, the risk factor of AD, is synthesized in the rough endoplasmic reticulum transferred through the Golgi complex to the cell membrane via vesicles with APP. APP is cleaved by α- or β-secretases, the latter of which produces soluble APP and C99. C99 is further cleaved into Aβ and AICD (APP-intracellular domain) by γ-secretase. Aβ fragments include Aβ40 and 42, the latter of which is much more prone to aggregation and more toxic to neurons than Aβ40. APP is also cleaved into soluble APP and C83 by α-secretase, and C-terminal fragment α (αCTF) is further processed by γ-secretase. APP-positive vesicles are transported to the cell membrane mostly through axons. In AD, disruption of proteolysis in AL, like in lysosomal storage diseases, is associated with neuronal autophagy pathology. A key genetic risk factor of AD, presenilin 1, has been shown to be required for lysosome acidification. Presenilin 1 mutations are known to be the most common cause of early onset familial AD.
Using five different APP-AD mouse models, Lee et al. have shown a new pathological pattern of autophagic stress they called “PANTHOS,” in individual neuronal perikarya; it involves massive perikaryal accumulation of poorly acidified autophagic vacuoles (Avs) with APP-βCTF/Aβ. For this, they use a neuron-specific transgenic mRFP-eGFP-LC3 probe of autophagy and pH, multiplex confocal imaging, and correlative light and electron microscopy (CLEM). In these animal models, they found decline of AL acidification in neurons before extracellular amyloid deposition. Acidification decreases in AL are associated with markedly lowered vATPase activity. Instead of acidified AL, they noticed the presence of partially acidified AL that increased size and number.
Although they demonstrated the presence of partially acidified AL, this situation of AL seems unique, and it remains unknown why pH homogenization does not occur in the luminal space. Accumulation of AVs with APP-bCTF/Ab suppressed vesicular transfer of APP from the ER-Golgi apparatus and the perinuclear networks of membrane tubules, in which fibrillar Aβ accumulates, and joined endosomes that further contributed as major sources of APP-βCTF/Aβ generation. Using a CLEM method, EM figures exhibited membrane blebbing; in the tip of blebs, many AVs were present and continuous to the perinuclear structures via the neck of the blebs.
Lee et al. further showed enlarged figures of PANTHOS in mice at higher age. By cytosolic and membrane/vesicle fraction analyses, they showed the presence of CtsB and CtsD in the cytosol of the brains from 6-month-old model mice but not in those from 2-month-old mice. As per immune-EM, positive signals for these enzymes were diffused in the cytosol in addition to AL/LY.
The mode of death of neurons with PANTHOS was caspase-3 independent. Although the authors showed the death mode is non-apoptotic (caspes-3 independent), membrane blebbing that is thought to be representative of apoptotic death occurs in PANTHOS neurons. Considering the promotion of lysosomal membrane permeabilization and cathepsin release into the cytosol, the death mode may be linked to necrosis or autophagy. Anyhow, since the death mode of PANTHOS neurons is quite unique in its features, PANTHOS neuron death may be a new mode of cell death due to strong autophagy stress.
The authors further showed that these PANTHOS figures develop amyloid (senile) plaques with extracellular accumulation of APP-bCTF/Aβ in these model mice. They also showed the presence of PANTHOS neurons in human AD degenerating brains with Braak II stage.
NOVA Medical School
This new paper throws new light onto the longstanding question of what is the origin of amyloid plaques. The authors use a new methodological approach, a transgenic mouse model overexpressing in neurons LC3 tagged with two fluorescent proteins, GFP and RFP, being GFP-fluorescence-sensitive to acidic pH, allowing for reporting on the acidification of autophagosomes upon fusion with lysosomes or endolysosomes. The authors detected GFP accumulation upon inducing lysosome dysfunction in vivo. This technological advancement allowed them to inquire whether and when lysosomal dysfunction occurs in AD, using five different models based on overexpression of APP with other mutations alone, in combination with presenilin1 mutants, or alone. The authors find that acidification defects occur early in neurons that accumulate Aβ, before apparent neurodegeneration.
These results confirm:
In the future, it will be necessary to experimentally induce/revert what is cause or consequence in the sequence of events, intraneuronal amyloid, lysosome dysfunction, in plaque formation, to determine the origin of amyloid plaques, especially in the most common forms of AD that develop in the absence of the familial mutations.
References:
Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Abeta42 accumulation in human brain. Am J Pathol. 2000 Jan;156(1):15-20. PubMed.
Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005 Oct;26(9):1235-44. PubMed.
Almeida CG, Takahashi RH, Gouras GK. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J Neurosci. 2006 Apr 19;26(16):4277-88. PubMed.
Schützmann MP, Hasecke F, Bachmann S, Zielinski M, Hänsch S, Schröder GF, Zempel H, Hoyer W. Endo-lysosomal Aβ concentration and pH trigger formation of Aβ oligomers that potently induce Tau missorting. Nat Commun. 2021 Jul 30;12(1):4634. PubMed.
University of California, Irvine
This interesting and exciting paper by Nixon and co-workers confirms and extends a model for neuritic plaque formation that we previously reported (see Fig 4 below from Pensalfini et al., 2014).
We used a fibril-specific monoclonal antibody, mOC78, which recognizes a unique discontinuous epitope, to show that neurons accumulate aggregated Aβ and APP-CTF in a perinuclear and nuclear location, ultimately leading to the death of neurons and deposition as “neuritic” plaques. The center of these plaques contains neuronal chromatin marked by NeuN immunoreactivity.
We also showed that neurons with perinuclear and nuclear amyloid are found in human brain and are elevated in plaque stage A and B, suggesting that they are early events in plaque pathology. In subsequent work, we showed that early treatment of 5XFAD mice with a CSF1R inhibitor to ablate microglia at 2 months (prior to neuritic plaque deposition) blocked intraneuronal amyloid accumulation (Step 1 in Fig 4) and also the subsequent deposition of neuritic plaques (Step 4) by approximately 90 percent, suggesting that there is a precursor–product relationship between intraneuronal amyloid and neuritic plaques (Sosna et al., 2018).
The intraneuronal amyloid model of the amyloid hypothesis has important implications for therapeutic development. Gamma-secretase and BACE1 inhibitors have been developed to block the secretion of soluble, monomeric Aβ. In clinical trials, these drugs actually caused a worsening of cognition compared to placebo. Inhibiting APP-CTF and APP cleavage would be expected to cause an increase in the lifetime and concentrations of these precursor substrates, which could favor intraneuronal amyloid aggregation and accumulation. It would be interesting to know if these drugs affect intraneuronal amyloid and if drugs could be found to increase the secretion of soluble monomeric Aβ. In addition, monoclonal antibodies that bind to and remove plaques have been approved by the FDA. If neuritic plaques are derived from dying neurons that have accumulated intraneuronal amyloid, then they really are “tombstone markers” of antecedent pathology and removing them is unlikely to raise the dead. Perhaps antibodies that target oligomers and do not bind to plaques would be more effective by blocking the seeding of more intraneuronal amyloid accumulation.
References:
Pensalfini A, Albay R 3rd, Rasool S, Wu JW, Hatami A, Arai H, Margol L, Milton S, Poon WW, Corrada MM, Kawas CH, Glabe CG. Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol Dis. 2014 Nov;71:53-61. Epub 2014 Aug 1 PubMed.
Sosna J, Philipp S, Albay R 3rd, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, Glabe CG. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer's disease. Mol Neurodegener. 2018 Mar 1;13(1):11. PubMed.
IIBB/CSIC/CIBERNED
This article represents a remarkable advance in the hypothesis that amyloid plaques are a late effect in the causal chain that leads to neurodegeneration. This study will help now to refocus research on the process of neuronal quality control of APP. It is remarkable that in endosomes, the authors also find CTF as well as Aβ.
The quality control of APP may happen in synaptic terminals and retro-transported endosomes accumulate in the soma. I am thrilled to learn that previous work from Dr. Gouras and others is finally vindicated with a great study that represents a leap forward.
TrueBinding
This is an interesting paper confirming the origin of amyloid plaques. We showed a similar result using the M78 monoclonal antibody, as my former supervisor Dr. Charles Glabe wrote in his comments (Pensalfini et al., 2014). M78 is an aggregation-specific, conformation-dependent monoclonal antibody raised against fibrillar Aβ42.
In 3xTg-AD mice, neuritic plaques at 12 months display the same spatial organization of centrally co-localized M78, diffuse chromatin, and neuronal nuclear NeuN staining surrounded by peripheral M78 and APP-CTF immunoreactivity as observed in neurons, indicating that neuritic plaques arise from degenerating neurons with intracellular amyloid immunoreactivity.
The same staining pattern was observed in neuritic plaques in human AD brains, showing elevated intracellular M78 immunoreactivity at intermediate stages of amyloid pathology (Braak A and B) compared to no amyloid pathology and late-stage amyloid pathology (Braak 0 and C, respectively).
These results indicate that intraneuronal protein aggregation and amyloid accumulation is an early event in AD, and that neuritic plaques are initiated by the degeneration and death of neurons by a mechanism that may be related to the formation of extracellular traps by neutrophils. M78 immunoreactivity co-localizes with Aβ, and APP carboxyl terminus (APP-CTF) immunoreactivity in perinuclear compartments at intermediate times in 10-month 3XTg-AD mice indicates that this represents misfolded and aggregated protein rather than normally folded APP.
References:
Pensalfini A, Albay R 3rd, Rasool S, Wu JW, Hatami A, Arai H, Margol L, Milton S, Poon WW, Corrada MM, Kawas CH, Glabe CG. Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol Dis. 2014 Nov;71:53-61. Epub 2014 Aug 1 PubMed.
Institut de Pharmacologie Moléculaire et Cellulaire
This great paper is a significant step toward our understanding of senile plaques genesis. It also strengthens the view that autolysosomal and autophagic defects occur upstream of plaque formation and could contribute to their formation. Overall, these observations agree with the fact that autophagic defects and endo-lysosomal alterations are early stigmata taking place not only in AD mouse models but also in human AD-affected brains.
This agrees well with our report on an interplay between the β-secretase-derived C99 fragment accumulation and endolysosomal and autophagic alterations. Thus, exacerbated accumulation of C99 triggers endolysosomal defects while, conversely, lysosomal alkalinization triggers C99 clearance reduction and thereby, its propensity to aggregate. Of note, these perturbations appeared independent of Aβ and thus likely occur before plaques appear. Indeed, they were enhanced upon γ-secretase inhibition (Lauritzen et al., 2016).
Thus, one could envision that there exists a dual contribution of C99 (our study) and intracellular Aβ (Nixon et al.'s work) in autophagic vacuoles. The respective contributions, and whether they could act synergistically to trigger PANTHOS, remain to be determined.
References:
Lauritzen I, Pardossi-Piquard R, Bourgeois A, Pagnotta S, Biferi MG, Barkats M, Lacor P, Klein W, Bauer C, Checler F. Intraneuronal aggregation of the β-CTF fragment of APP (C99) induces Aβ-independent lysosomal-autophagic pathology. Acta Neuropathol. 2016 Aug;132(2):257-76. Epub 2016 Apr 30 PubMed. Correction.
Tokyo Medical and Dental University
Prélude du tombeau—from PANTHOS to TRIAD?
The group of Ralph Nixon reported that poorly acidified autolysosomes accumulate intracellular Aβ, protrusions of cytoplasm filled with autophagosome are sectioned by microglia, and the phenotype is related to cell death.
This highly interesting work is extremely similar to the morphology of degenerative/dystrophic neurites described in the classic textbook Greenfield’s Neuropathology (see in image below). It is also surprisingly homologous to the morphology we reported (Fujita et al., 2016, see upper right, lower left below).
It is probably consistent with our previous results that intracellular Aβ, which was discovered by my group (Shoji et al., 2000) and reconfirmed by the group of Mark Smith (Zhu et al., 2003) causes TRIAD (transcriptional repression-induced atypical cell death) necrosis based on transcriptional repression, especially of YAP-TEAD mediated transcription (Hoshino et al., 2006; Mao et al., 2016), and that TRIAD necrosis of neurons releases DAMPs such as HMGB1 causing secondary neurite degeneration (Fujita et al., 2016; Tanaka et al., 2020), and secondary necrosis (Tanaka et al., 2021).
Upper left: Image of senile (neuritic) plaque obtained by electron microscopy, published in Greenfield’s Neuropathology (7th edition). Upper right: A single neuron death by TRIAD necrosis in 5xFAD mice (Fujita et al., Sci Rep 2016). The similar immunostains in postmortem brains of human AD patients and in brains of APP-KI mice were also published in the same paper. Lower left: Dystrophic neurite or PANTHOS? Structures surrounding extracellular Aβ deposit derived from a single neuron death, which are filled with autophagosomes. (Tanaka et al, Nature Commun 2021).
These findings forced us to consider that extracellular Aβ plaques might be actually the tombstones of intracellular Aβ after necrosis (Tanaka et al., 2020), and that this pathological process initiates the exponential expansion of neuronal death based on “TRIAD necrosis to TRIAD necrosis” far before the time point of extracellular Aβ plaque formation (Okazawa, 2021). This line of our idea would probably match with that of the authors.
Interestingly, our first paper in this line indicates that JNK, the key pathway suppressing autophagy (Xu et al., 2011), is activated when neurons stay viable. This fact might be consistent with TRIAD necrosis instead of autophagic cell death occurring in the center of the cell death foci, even though structures of either the same cell or different cells distal of the cell death are filled with autophagosomes.
In our live imaging of iPS cell-derived neurons with APP mutations, and in our EM images of postmortem brains of human MCI/AD patients, we observed vacuole formation including Aβ in the cytoplasm, and that these sometimes protrude like balloons (Tanaka et al., 2020) where no noticeable increase of autophagosomes is detected in the cytoplasm (Hoshino et al., 2006). The morphology of PANTHOS, which forms focal protrusions, could be consistent with TRIAD necrosis of Aβ-positive human neurons (Tanaka et al., 2020).
In TRIAD necrosis, the cytoplasmic protrusion is driven by enlargement of endoplasmic reticulum rather than accumulation of autophagosomes (Hoshino et al., 2006; Mao et al., 2016; Tanaka et al., 2020). The physical force of the autolysosome, a small membranous organelle, is too weak to deform the cell shape. Therefore, we wonder whether the structural change of PANTHOS might be secondary to ER enlargement.
Moreover, the Greenfield Neuropathology textbook contains an EM image of dystrophic neurites having obvious cell membranes and filled with autophagosomes that exist around extracellular Aβ plaque without cell membrane (Greenfield’s Neuropathology). On the other hand, PANTHOS reported here did not reveal obvious cell membranes, and the loss of the cell membrane is plausible when the cell is dying. Therefore, we wonder whether PANTHOS referred to in this article is distinct from dystrophic neurites coming from the other neurons and surrounding Aβ plaques described in the classic textbook Greenfield’s Neuropathology and observed by our group (Fujita et al., 2016; Tanaka et al., 2020). Actually, in Figure 6d of the Nixon group paper, they showed dystrophic neurites (DN) that included gold particles for immuno-electron microscopy.
Finally, we wonder about the relationship between PANTHOS and TRIAD (Hoshino et al., 2006). Could PANTHOS be an earlier process of TRIAD, the actual cell death of neurons possessing intracellular Aβ (Tanaka et al., 2020)?
—Kyota Fujita, Hidenori Homma, and Hikari Tanaka of the Department of Neuropathology, Tokyo Medical and Dental University, are co-authors of this comment.
References:
Graham DI, Lantos PL. Greenfield’s Neuropathology (2 Vol. Set). 7th Ed. vol. 2, 210
Fujita K, Motoki K, Tagawa K, Chen X, Hama H, Nakajima K, Homma H, Tamura T, Watanabe H, Katsuno M, Matsumi C, Kajikawa M, Saito T, Saido T, Sobue G, Miyawaki A, Okazawa H. HMGB1, a pathogenic molecule that induces neurite degeneration via TLR4-MARCKS, is a potential therapeutic target for Alzheimer's disease. Sci Rep. 2016 Aug 25;6:31895. PubMed.
Shoji M, Iwakami N, Takeuchi S, Waragai M, Suzuki M, Kanazawa I, Lippa CF, Ono S, Okazawa H. JNK activation is associated with intracellular beta-amyloid accumulation. Brain Res Mol Brain Res. 2000 Dec 28;85(1-2):221-33. PubMed.
Zhu X, Ogawa O, Wang Y, Perry G, Smith MA. JKK1, an upstream activator of JNK/SAPK, is activated in Alzheimer's disease. J Neurochem. 2003 Apr;85(1):87-93. PubMed.
Hoshino M, Qi ML, Yoshimura N, Miyashita T, Tagawa K, Wada Y, Enokido Y, Marubuchi S, Harjes P, Arai N, Oyanagi K, Blandino G, Sudol M, Rich T, Kanazawa I, Wanker EE, Saitoe M, Okazawa H. Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of YAP isoforms and p73. J Cell Biol. 2006 Feb 13;172(4):589-604. PubMed.
Mao Y, Chen X, Xu M, Fujita K, Motoki K, Sasabe T, Homma H, Murata M, Tagawa K, Tamura T, Kaye J, Finkbeiner S, Blandino G, Sudol M, Okazawa H. Targeting TEAD/YAP-transcription-dependent necrosis, TRIAD, ameliorates Huntington's disease pathology. Hum Mol Genet. 2016 Sep 12; PubMed.
Tanaka H, Homma H, Fujita K, Kondo K, Yamada S, Jin X, Waragai M, Ohtomo G, Iwata A, Tagawa K, Atsuta N, Katsuno M, Tomita N, Furukawa K, Saito Y, Saito T, Ichise A, Shibata S, Arai H, Saido T, Sudol M, Muramatsu SI, Okano H, Mufson EJ, Sobue G, Murayama S, Okazawa H. YAP-dependent necrosis occurs in early stages of Alzheimer's disease and regulates mouse model pathology. Nat Commun. 2020 Jan 24;11(1):507. PubMed.
Tanaka H, Kondo K, Fujita K, Homma H, Tagawa K, Jin X, Jin M, Yoshioka Y, Takayama S, Masuda H, Tokuyama R, Nakazaki Y, Saito T, Saido T, Murayama S, Ikura T, Ito N, Yamamori Y, Tomii K, Bianchi ME, Okazawa H. HMGB1 signaling phosphorylates Ku70 and impairs DNA damage repair in Alzheimer's disease pathology. Commun Biol. 2021 Oct 11;4(1):1175. PubMed.
Okazawa H. Intracellular amyloid hypothesis for ultra-early phase pathology of Alzheimer's disease. Neuropathology. 2021 Apr;41(2):93-98. PubMed.
Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011 Feb 15;25(4):310-22. PubMed.
View all comments by Hitoshi OkazawaNathan Kline Institute/NYU Langone Med
New York University School of Medicine/Nathan Kline Institute
We are grateful to all investigators who commented on our study. We greatly appreciate their past contributions to this evolving topic and their new insights. The detailed commentary and proposal by Dr. Okazawa and colleagues especially are intriguing, and below we offer our thoughts on the comparison between TRIAD and PANTHOS-related mechanisms in AD.
We conclude that TRIAD and PANTHOS are distinct cell-death trajectories. This is consistent with evidence for a multiplicity of cell-death mechanisms known in different neuronal populations (Yang et al, 2008), or even in the same population at different stages of neuronal compromise (Nixon and Yang, 2012). That said, it is possible that certain aspects of TRIAD and PANTHOS may overlap. For example, lysosomal dysfunction, such as autophagic flux suppression mediated by vATPase inhibition, promotes YAP accumulation in cardiomyocytes (Ikeda et al., 2021).
Although PANTHOS-related lysosomal death of neurons and transition to senile plaques is a significant aspect of our report, the most important finding of our study, in our opinion, is the very early onset of lysosomal dysfunction in substantial percentages of neurons in vulnerable populations. In a separate study (Im et al., 2022), defective lysosomal acidification can be traced to accumulation of APP-βCTF and their inhibitory interactions with vATPase. These findings position lysosome dysfunction at the same proximal (causal) stage as proposed for Aβ, emphasizing the critical partnership between genetically driven lysosomal dysfunction and APP (Nixon, 2017). PANTHOS can also be demonstrated in AD brain, suggesting that additional genetic and environmental factors may drive a similar process in “sporadic” AD.
View all comments by Ralph NixonMake a Comment
To make a comment you must login or register.