APP C-Terminal Fragments Stifle Calcium Flow from ER to Lysosomes
Quick Links
C-terminal fragments of amyloid precursor protein have been blamed for endolysosomal dysfunction in Alzheimer’s disease. Now, scientists led by Wim Annaert at KU Leuven, Belgium, claim that’s because these fragments disrupt calcium flux. In the April 15 Developmental Cell, they reported that knocking out presenilin 1 and 2 in mouse fibroblasts causes APP-CTFs to accumulate in lysosomes, especially at junctions with the ER. This prevents the flow of calcium ions between the two compartments, slowing endolysosomal maturation. Reducing the APP-CTFs restored lysosome function.
- APP CTFs accumulate in lysosomes, especially at junctions with the endoplasmic reticulum.
- This stunts calcium flow from the ER to lysosomes, stalling the cellular waste disposal system.
- Knocking out APP normalizes endolysosome function.
Scientists praised the research. “This is beautiful cell biology work addressing important questions,” Gunnar Gouras of Lund University, Sweden, told Alzforum. “This study provides exciting mechanistic insights into how APP-CTFs, and not Aβ, may contribute to the lysosomal dysfunction in Alzheimer’s disease,” wrote Stefan Lichtenthaler at the German Center for Neurodegenerative Diseases, Munich (comments below). Aβ has previously been tied to bloated, neurotoxic lysosomes (Jun 2022 news).
APP-CTFs also have been incriminated in lysosome malfunction. Scientists led by Marc Tessier-Lavigne of Stanford University found that familial AD mutations in APP or presenilin 1 caused endosomal dysfunction due to accumulation of β-CTFs (Aug 2019 news). Randy Nixon at New York University in Orangeburg reported that β-CTFs, created when β-secretase cleaves APP, block the assembly of vacuolar ATPase that fuels lysosome acidification and that reducing β-CTF production revived the organelles (Jul 2023 news). Others have reported poor acidification caused by presenilin mutations as well (Chou et al., 2023; Martin-Maestro et al., 2017; see Nixon comment below).
Annaert has long been a proponent of a different reason for β-CTF lysosomal dysfunction: calcium imbalance. He has reported that presenilins, better known as catalytic components of γ-secretase, also regulate lysosomal calcium levels (Jul 2012 news). Without presenilin, calcium levels in the lysosome falls and the cellular waste system skids to a halt. In the latest twist, he implicates APP-CTFs in this signaling snafu.
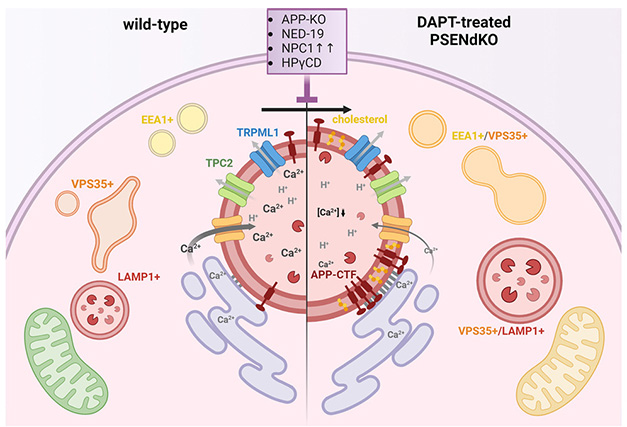
With or Without Presenilin. In healthy cells (left), calcium flows from the endoplasmic reticulum (purple) to the lysosomes (red circle in center). Early endosomes (EEA1+), recycling endosomes (VPS35+), and late endosomes/lysosomes (LAMP1+) function normally. When APP-CTFs accumulate in the lysosomal membrane (right), lysosomal calcium drops and endolysosomes swell. Mitochondria (green, bottom left) made less ATP (yellow, bottom right) though it’s unclear why. [Courtesy of Bretou et al., Developmental Cell, 2024.]
To better understand the role of presenilins in the endolysosomal system, first author Marine Bretou turned to mouse embryonic fibroblasts. “We wanted to work with a simple model, then validate findings in neurons later,” Annaert told Alzforum.
First, Bretou treated the fibroblasts with the γ-secretase inhibitor DAPT to prevent proteolytic processing of APP-CTFs. Within a day, the fragments had accumulated in lysosomes and late endosomes, which had swollen. By days three and four, recycling endosomes and early endosomes, respectively, had enlarged as well. These results indicate a delay in endosome-to-lysosome maturation, with lysosomes failing first.
Calcium levels in lysosomes also fell by 30 percent in the 24 hours after DAPT. Bretou saw the same effects of DAPT in primary mouse hippocampal neurons: APP-CTF accumulation, lysosomal calcium decrease, and endolysosomal swelling.
To tease apart APP-CTF’s from Aβ’s role in endolysosomal demise, Bretou and colleagues knocked out presenilin 1 and 2 in mouse embryonic fibroblasts to completely eliminate Aβ production since some still occurs with DAPT treatment. In these knockout cells, lysosomes had about fivefold less Ca2+ than did lysosomes in wild-type fibroblasts.
This imbalance did not stem from calcium leaking out of lysosomes but from restricted calcium transport into lysosomes from the endoplasmic reticulum, the authors found. When Bretou emptied lysosomes of Ca2+ using the dipeptide gly-phe-β-naphtylamide to disrupt their membranes, those in control cells refilled quickly, while lysosomes in presenilin knockout cells did not. Control lysosomes failed to refill when Bretou added an endoplasmic reticulum Ca2+ pump inhibitor to the medium. The ER pump recycles cytosolic calcium back into the organelle. Because the lysosomes did not refill with calcium, either, the authors concluded that the ER was the source of these ions.
To directly test whether CTFs are involved in lysosomal Ca2+ flux, the scientists knocked out APP and both presenilins in fibroblasts, then reintroduced either α- or β-CTFs, fragments cleaved by α- and β-secretase, respectively. To their surprise, they found that both equally reduced lysosomal calcium and stunted endolysosomal function. Previous work suggested only the shorter β-CTFs disrupted lysosome function.
Further support for the idea that CTFs dysregulate calcium flow came from super-resolution microscopy. It showed the fragments clustering on the lysosomal membrane, right where it contacts the ER (image below). Scientists believe the ER exchanges small molecules and ions, such as Ca2+, with organelles through these membrane junctions (reviewed by Burgoyne et al., 2015). The authors suspect the CTFs keep the lysosomes tightly bound to the ER, as well, because in PS1/2 knockout lines, the lysosomes moved sluggishly around the cytoplasm.

Cozy Organelles. When APP β-CTFs were added to mouse fibroblasts lacking presenilin 1/2 and APP, the fragments (red) bound to lysosomes (blue), which swelled and became enmeshed in the endoplasmic reticulum (green). [Courtesy of Bretou et al., Developmental Cell, 2024.]
Could the stalled endolysosomes be jolted back into action? Indeed, expressing human presenilin 1 in the presenilin double knockout fibroblasts shrank endosomes and lysosomes, enabled lysosomes to move normally through the cytoplasm, and normalized lysosomal Ca2+ levels. Deleting APP in the presenilin knockout fibroblasts also ameliorated endolysosomal defects, directly implicating the precursor protein in weakening this recycling system.
These endolysosomal deficits also occurred when the triple knockout fibroblasts expressed the APP intracellular domain (AICD) carrying an N-terminal motif prone to modification with myristic acid. The lipid anchors AICD to the membrane, effectively turning it into a CTF. AICD contains the YENPTY sequence, to which multiple proteins bind, including Abl kinase and the sortilin receptor SorLA. When the researchers mutated the motif to abolish these interactions, lysosomal calcium levels and endolysosome morphology were normal. To the authors, this suggested that YENPTY interactions, in addition to accumulation of APP-CTFs, induces endolysosomal dysfunction.
As a corollary of this, γ-secretase, which releases AICD into the cytoplasm, would protect against endolysosomal defects. AICD, like the Notch intracellular domain, is believed to form complexes that regulate gene expression, but the authors think their findings dispel that notion. They claim that failure to degrade CTFs, not production of AICD, sustains downstream signaling, instigating lysosomal dysfunction. Rick Livesey, University College London, thinks this is plausible. “Proteolysis of APP-CTFs is an off switch for APP-CTF-mediated functions, rather than activating downstream signaling,” he wrote (comment below). “In this scenario, accumulation of APP-CTFs is effectively a toxic gain-of-function.”
On that note, the data offer another explanation for why γ-secretase inhibitors failed in clinical studies in AD with one, semagacestat, worsening cognition (Doody et al., 2013). “It is tempting to hypothesize that [this] effect on cognition might be mediated by APP-CTFs and/or endolysosomal dysfunction,” wrote Rik van der Kant of Vrije University in Amsterdam (comment below). As such, the findings support the use of γ-secretase modulators to encourage processing of APP-CTFs into shorter, non-toxic Aβ peptides or BACE inhibitors to prevent production of β-CTFs to begin with (Jul 2023 conference news; Aug 2022 conference news).—Chelsea Weidman Burke
References
News Citations
- Behold PANTHOS, a Toxic Wreath of Perinuclear Aβ That Kills Neurons
- Familial AD Mutations, β-CTF, Spell Trouble for Endosomes
- Too Basic: APP β-CTF's YENTPY Motif Binds Proton Pump, Thwarts Lysosomes
- Presenilins and Calcium: A Lysosomal Stew With Acid Controversy
- Give BACE Inhibitors a Second Chance?
- Can BACE Inhibitors Stage a Comeback?
Paper Citations
- Chou CC, Vest R, Prado MA, Wilson-Grady J, Paulo JA, Shibuya Y, Moran-Losada P, Lee TT, Luo J, Gygi SP, Kelly JW, Finley D, Wernig M, Wyss-Coray T, Frydman J. Proteostasis and lysosomal quality control deficits in Alzheimer's disease neurons. bioRxiv. 2023 Mar 27; PubMed.
- Martín-Maestro P, Gargini R, A Sproul A, García E, Antón LC, Noggle S, Arancio O, Avila J, García-Escudero V. Mitophagy Failure in Fibroblasts and iPSC-Derived Neurons of Alzheimer's Disease-Associated Presenilin 1 Mutation. Front Mol Neurosci. 2017;10:291. Epub 2017 Sep 14 PubMed.
- Burgoyne T, Patel S, Eden ER. Calcium signaling at ER membrane contact sites. Biochim Biophys Acta. 2015 Sep;1853(9):2012-7. Epub 2015 Feb 4 PubMed.
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, , Siemers E, Sethuraman G, Mohs R. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013 Jul 25;369(4):341-50. PubMed.
Further Reading
Primary Papers
- Bretou M, Sannerud R, Escamilla-Ayala A, Leroy T, Vrancx C, Van Acker ZP, Perdok A, Vermeire W, Vorsters I, Van Keymolen S, Maxson M, Pavie B, Wierda K, Eskelinen EL, Annaert W. Accumulation of APP C-terminal fragments causes endolysosomal dysfunction through the dysregulation of late endosome to lysosome-ER contact sites. Dev Cell. 2024 Jun 17;59(12):1571-1592.e9. Epub 2024 Apr 15 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.