Acetylated Tau Mucks Up Memories
Quick Links
Toxic tau protein, untangled but donning a pair of acetyl groups, infiltrates synapses and prevents memory formation, according to a paper in the March 31 Neuron online. The study explains one way tau can interfere with cognition, say the researchers, led by senior author Li Gan at the Gladstone Institute of Neurological Disease in San Francisco. They traced weakening of neural plasticity to a crucial synaptic protein known as KIBRA, which disappears when acetylated tau arrives in the synapse. The authors claim KIBRA is the “missing link” between tau and memory loss.
“This is a really phenomenal paper,” commented Todd Cohen of the University of North Carolina in Chapel Hill, who was not involved in the study. “We have to consider acetylation as a dominant player in controlling tau function at the synapse, and possibly elsewhere.”
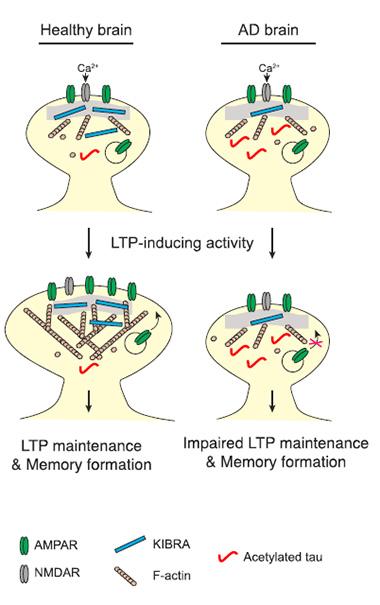
Synapse machinery
KIBRA recruits actin and AMPA receptors to strengthen synapses. In AD, acetylated tau may suppress KIBRA, impairing memory. [Courtesy of Neuron, Tracy et al.]
Hyperphosphorylated tau has long been known to accumulate and aggregate in the brains of people with Alzheimer’s or frontotemporal dementia. Over the last few years, however, a handful of scientists have turned their attention to a different tau modification—acetylation. The acetyltransferase p300 attaches acetyl groups to lysine residues in tau. Upward of 20 tau lysines may be subject to such modification, though Gan said the precise number is uncertain. Both histone deacetylase 6, SIRT1, and potentially other deacetylases can remove the groups. In 2010, Gan and colleagues reported that acetylation protects tau from degradation, allowing hyperphosphorylated tau to accumulate (see Sep 2010 news on Min et al., 2010).
To identify acetylation sites relevant to disease, the authors used mass spectrometry to analyze tau from the brains of people who died of Alzheimer’s. They found acetyl groups at lysines 174, 274, and 281. In 2015, the Gan lab reported that a version of tau with a glutamine in place of lysine-174, which mimicked the acetylated tau, caused hippocampal atrophy and memory defects in mice (see Sep 2015 news). In the new paper in Neuron, first author Tara Tracy and colleagues focused on the other two sites, which occur in the protein’s microtubule-binding domain.
Tracy generated transgenic mice (tauKQ) that expressed human tau with glutamine residues at positions 274 and 281. Another line, called tauKQ-high, expressed about 20 percent more of this construct. The tauKQs got phosphorylated, but did not form tangles.
Both tauKQ mice acted fairly normally, and lived full lifespans. However, in a water maze, tauKQ-high mice failed to recall the location of a hidden platform, indicating they had difficulty with spatial memory. Tracy also assessed whether the tauKQ-high mice failed to form or distinguish two similar memories, as people with mild cognitive impairment or AD are thought to do (Ally et al., 2013; Wesnes et al., 2014; Yassa et al., 2010). She tested memory in a fear-conditioning experiment that utilized two very similar contexts (Nakashiba et al., 2012; McHugh et al., 2007). She trained the mice in two identical cages that had different environmental cues. One smelled like Windex and had background noise from a fan. The other smelled like Simple Green and contained a solid black, tent-like cover inside. Only in the noisy Windex cage did the mice receive a foot shock. After a learning period, nontransgenic mice came to recognize that the Simple Green cage was safe, and were less likely to freeze in anticipation of a shock when placed in it. In contrast, TauKQ-high mice froze in both cages, indicating they had trouble differentiating the two.
Tracy found a potential explanation for these memory deficits when she measured long-term potentiation, which is needed for synaptic remodeling, in hippocampal slices. When she stimulated neurons in the dentate gyrus—the part of the hippocampus responsible for distinguishing memories of similar objects or environments—their output stayed high for more than an hour in slices from nontransgenic mice and mice expressing wild-type human tau. In the tauKQ lines, the output dropped faster, indicating the synapse was unable to maintain strength.
The authors suspected this defect resulted from failure to deliver AMPA receptors to the spine tips, as normal dendrites do. Tracy tested this in hippocampal rat neurons expressing tauKQ or wild-type tau. On applying glycine to stimulate long-term potentiation, the tauWT neurons polymerized actin, which is needed for receptor transport, and dispatched AMPA receptors to the spine surfaces. The tauKQ neurons did neither. Going back to the mouse hippocampal slices, Tracy treated them with jasplakinolide, which polymerizes actin. This restored the long-term potentiation of tauKQ-high slices to normal, suggesting acetylated tau acts upstream of actin polymerization to disrupt synaptic plasticity.
Next, Tracy and Gan wondered what molecule might link tau to actin polymerization. They considered the kidney/brain postsynaptic protein, KIBRA. It interacts with AMPA receptors and proteins that regulate actin (Duning et al., 2008; Kremerskothen et al., 2003; Kremerskothen et al., 2005). In genetic studies, KIBRA has been linked to memory performance in cognitively healthy people (see Oct 2006 news) as well as late-onset Alzheimer’s (Rodríguez-Rodríguez et al., 2009; Corneveaux et al., 2010; Burgess et al., 2011). Mice lacking KIBRA have impaired long-term potentiation and memory (see Oct 2011 news). Tracy and Gan discovered less KIBRA in brain homogenates from people who died of AD, compared with non-demented control brains.
To test whether KIBRA participated in the synaptic plasticity pathway that tau interfered with, Tracy returned to the cultured neurons expressing tauKQ. Co-expressing excess KIBRA restored their ability to polymerize actin and traffic AMPA receptors. While they have not yet worked out how, the authors present a model whereby acetylated tau causes a drop in KIBRA at the synapse. Their experiments indicate that the loss of KIBRA leads to poor actin polymerization and AMPA receptor transport, and impaired synaptic plasticity (see image above).
This is just one of many ways tau could be toxic, noted scientists who spoke with Alzforum. “I would not be surprised if there are other actions of acetylated tau,” commented Li-Huei Tsai of the Massachusetts Institute of Technology, who did not participate in the work. She wondered if acetylation might also affect toxic actions such as the seeding of tau aggregates or its spread between cells. The combination of acetylation, phosphorylation, and aggregation likely works in concert to make tau dangerous, Tsai said. In fact, Cohen’s work suggests one such interaction; acetylation promotes tau fibril formation, at least in vitro (see Mar 2011 news).
Cohen noted that tau can be phosphorylated at 40-some sites, acetylated at another couple of dozen, and is subject to other modifications such as glycosylation and methylation too (see Jul 2015 news; Song et al., 2015). “We may have a very complex code of modifications,” he said. Studying how those different modifications work together—rather than just as one or two at a time, as Tracy did—will be important in future research, he said. While this will be complicated to do, he suggested computational or proteomics methods might help crack the code.
In the meantime, Gan and colleagues are already hoping to apply what they have learned about tau acetylation for therapeutics. The anti-inflammatory medication salsalate inhibits p300, the tau acetyltransferase, and Gan and Adam Boxer at the University of California in San Francisco are already testing it in people with the tauopathy progressive supranuclear palsy. Gan said she is interested in trying salsalate in people with Alzheimer’s, too, even as she works to identify compounds that would more potently and specifically inhibit p300. The new work suggests that if scientists could find small molecules that enhance KIBRA function, they too might have clinical potential. Tsai pointed out enhancing SIRT1’s ability to remove acetyl groups might also be beneficial.—Amber Dance
References
Alzpedia Citations
News Citations
- Tau Timing: New Findings on Disease Progression, Clearance
- New Type of Toxic Tau? Acetylated Form Correlates With Memory Defects
- Thanks for the Memories—KIBRA Alleles Predict Top Memory Performers
- Keys Found to KIBRA Memory Mystery
- Tau Modification—Move Over Phosphate, Make Room for Acetylation
- Inventory of Tau Modifications Hints at Undiscovered Functions
Paper Citations
- Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010 Sep 23;67(6):953-66. PubMed.
- Ally BA, Hussey EP, Ko PC, Molitor RJ. Pattern separation and pattern completion in Alzheimer's disease: Evidence of rapid forgetting in amnestic mild cognitive impairment. Hippocampus. 2013 Jun 27; PubMed.
- Wesnes KA, Annas P, Basun H, Edgar C, Blennow K. Performance on a pattern separation task by Alzheimer's patients shows possible links between disrupted dentate gyrus activity and apolipoprotein E ∈4 status and cerebrospinal fluid amyloid-β42 levels. Alzheimers Res Ther. 2014;6(2):20. Epub 2014 Apr 15 PubMed.
- Yassa MA, Stark SM, Bakker A, Albert MS, Gallagher M, Stark CE. High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. Neuroimage. 2010 Jul 1;51(3):1242-52. PubMed.
- Nakashiba T, Cushman JD, Pelkey KA, Renaudineau S, Buhl DL, McHugh TJ, Rodriguez Barrera V, Chittajallu R, Iwamoto KS, McBain CJ, Fanselow MS, Tonegawa S. Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell. 2012 Mar 30;149(1):188-201. Epub 2012 Feb 23 PubMed.
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK, Lowell BB, Fanselow MS, Wilson MA, Tonegawa S. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science. 2007 Jul 6;317(5834):94-9. Epub 2007 Jun 7 PubMed.
- Duning K, Schurek EM, Schlüter M, Bayer M, Reinhardt HC, Schwab A, Schaefer L, Benzing T, Schermer B, Saleem MA, Huber TB, Bachmann S, Kremerskothen J, Weide T, Pavenstädt H. KIBRA modulates directional migration of podocytes. J Am Soc Nephrol. 2008 Oct;19(10):1891-903. Epub 2008 Jul 2 PubMed.
- Kremerskothen J, Plaas C, Büther K, Finger I, Veltel S, Matanis T, Liedtke T, Barnekow A. Characterization of KIBRA, a novel WW domain-containing protein. Biochem Biophys Res Commun. 2003 Jan 24;300(4):862-7. PubMed.
- Kremerskothen J, Plaas C, Kindler S, Frotscher M, Barnekow A. Synaptopodin, a molecule involved in the formation of the dendritic spine apparatus, is a dual actin/alpha-actinin binding protein. J Neurochem. 2005 Feb;92(3):597-606. PubMed.
- Rodríguez-Rodríguez E, Infante J, Llorca J, Mateo I, Sánchez-Quintana C, García-Gorostiaga I, Sánchez-Juan P, Berciano J, Combarros O. Age-dependent association of KIBRA genetic variation and Alzheimer's disease risk. Neurobiol Aging. 2009 Feb;30(2):322-4. PubMed.
- Corneveaux JJ, Liang WS, Reiman EM, Webster JA, Myers AJ, Zismann VL, Joshipura KD, Pearson JV, Hu-Lince D, Craig DW, Coon KD, Dunckley T, Bandy D, Lee W, Chen K, Beach TG, Mastroeni D, Grover A, Ravid R, Sando SB, Aasly JO, Heun R, Jessen F, Kölsch H, Rogers J, Hutton ML, Melquist S, Petersen RC, Alexander GE, Caselli RJ, Papassotiropoulos A, Stephan DA, Huentelman MJ. Evidence for an association between KIBRA and late-onset Alzheimer's disease. Neurobiol Aging. 2010 Jun;31(6):901-9. PubMed.
- Burgess JD, Pedraza O, Graff-Radford NR, Hirpa M, Zou F, Miles R, Nguyen T, Li M, Lucas JA, Ivnik RJ, Crook J, Pankratz VS, Dickson DW, Petersen RC, Younkin SG, Ertekin-Taner N. Association of common KIBRA variants with episodic memory and AD risk. Neurobiol Aging. 2011 Mar;32(3):557.e1-9. PubMed.
- Song L, Lu SX, Ouyang X, Melchor J, Lee J, Terracina G, Wang X, Hyde L, Hess JF, Parker EM, Zhang L. Analysis of tau post-translational modifications in rTg4510 mice, a model of tau pathology. Mol Neurodegener. 2015 Mar 26;10:14. PubMed.
Further Reading
Papers
- Gorsky MK, Burnouf S, Dols J, Mandelkow E, Partridge L. Acetylation mimic of lysine 280 exacerbates human Tau neurotoxicity in vivo. Sci Rep. 2016 Mar 4;6:22685. PubMed.
- Zempel H, Mandelkow E. Lost after translation: missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014 Dec;37(12):721-32. Epub 2014 Sep 12 PubMed.
- Vogt-Eisele A, Krüger C, Duning K, Weber D, Spoelgen R, Pitzer C, Plaas C, Eisenhardt G, Meyer A, Vogt G, Krieger M, Handwerker E, Wennmann DO, Weide T, Skryabin BV, Klugmann M, Pavenstädt H, Huentelmann MJ, Kremerskothen J, Schneider A. KIBRA (KIdney/BRAin protein) regulates learning and memory and stabilizes Protein kinase Mζ. J Neurochem. 2013 Oct 7; PubMed.
- Boraxbekk CJ, Ames D, Kochan NA, Lee T, Thalamuthu A, Wen W, Armstrong NJ, Kwok JB, Schofield PR, Reppermund S, Wright MJ, Trollor JN, Brodaty H, Sachdev P, Mather KA. Investigating the influence of KIBRA and CLSTN2 genetic polymorphisms on cross-sectional and longitudinal measures of memory performance and hippocampal volume in older individuals. Neuropsychologia. 2015 Nov;78:10-7. Epub 2015 Sep 28 PubMed.
- Papenberg G, Salami A, Persson J, Lindenberger U, Bäckman L. Genetics and functional imaging: effects of APOE, BDNF, COMT, and KIBRA in aging. Neuropsychol Rev. 2015 Mar;25(1):47-62. Epub 2015 Feb 10 PubMed.
- Kauppi K, Nilsson LG, Adolfsson R, Eriksson E, Nyberg L. KIBRA polymorphism is related to enhanced memory and elevated hippocampal processing. J Neurosci. 2011 Oct 5;31(40):14218-22. PubMed.
- Almeida OP, Schwab SG, Lautenschlager NT, Morar B, Greenop KR, Flicker L, Wildenauer D. KIBRA genetic polymorphism influences episodic memory in later life, but does not increase the risk of mild cognitive impairment. J Cell Mol Med. 2008 Sep-Oct;12(5A):1672-6. PubMed.
News
- Tau Sullies Synaptic Function
- Tau’s Synaptic Hats: Regulating Activity, Disrupting Communication
- Not All About Dendrites: Presynaptic Tau Harms Plasticity, Too
- Toxic Tau Quiets Neuronal Networks
- Does Dendritic Tau Promote Plasticity?
- In Adult Mice, Reduced Tau Quiets Agitated Neurons
- Honolulu: The Missing Link? Tau Mediates Aβ Toxicity at Synapse
- APP Mice: Losing Tau Solves Their Memory Problems
Primary Papers
- Tracy TE, Sohn PD, Minami SS, Wang C, Min SW, Li Y, Zhou Y, Le D, Lo I, Ponnusamy R, Cong X, Schilling B, Ellerby LM, Huganir RL, Gan L. Acetylated Tau Obstructs KIBRA-Mediated Signaling in Synaptic Plasticity and Promotes Tauopathy-Related Memory Loss. Neuron. 2016 Apr 20;90(2):245-60. Epub 2016 Mar 31 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Universidad Autónoma de Madird
Universidad Autónoma de Madrid
Tau is mainly an axonal protein. However, it is also present at dendritic spines and it may play a toxic role there in Alzheimer’s disease and other tauopathies. That toxic role could be the consequence of tau modifications, such asphosphorylation, truncation, or acetylation. The role of acetylated tau in tauopathies is increasingly better understood. Tracy et al. describe, in an elegant manner, a possible mechanism for the role of acetylated tau (at lysines 274 and 281) in memory impairment using a mouse model overexpressing human tau with lysine to glutamine mutations to mimic lysine acetylation. Acetylated tau reduced the levels of kidney- and brain-expressed protein (KIBRA), a protein enriched in the postsynaptic density, and associated with late-onset sporadic AD. The mouse model data agreed with that finding, using AD patient samples. Thus, reduced KIBRA levels in AD patients with severe dementia was associated with enhanced acetylation of tau. Interestingly, increasing KIBRA levels in cultured hippocampal neurons from the transgenic mice corrected observed tau-mediated deficits in synaptic plasticity.
As the authors suggest, the next steps will be to determine precisely how acetylated tau decreases KIBRA levels, and more precisely clarify whether or not direct binding between acetylated tau and KIBRA exists, as has been previously described for tau and fyn kinase in dendrites (Ittner et al., 2010). The role played by actin, while analyzed in this manuscript, should also be studied in more depth. Recently, it was demonstrated using an optogenic approach that strengthening neuronal connections restores spine density and long-term memory in a PS1/APP AD mouse model (Roy et al., 2016). It would be interesting to see if the same occurs in this AD model based on overexpressing pseudoacetylated tau.
References:
Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010 Aug 6;142(3):387-97. Epub 2010 Jul 22 PubMed.
Roy DS, Arons A, Mitchell TI, Pignatelli M, Ryan TJ, Tonegawa S. Memory retrieval by activating engram cells in mouse models of early Alzheimer's disease. Nature. 2016 Mar 24;531(7595):508-12. Epub 2016 Mar 16 PubMed.
CogWellin Pharmaceuticals LLC
Finding acetylated tau at K-174 or 274 or 280 and/or at 281 in the CSF of AD patients versus control subjects free of cognitive issues will add more credence to the theory that acetylated tau is the missing link. If one can find such acetylated tau (which are at the microtubular domain binding region of tau) in the CSF of humans with AD and not in healthy controls, it will be a very important finding furthering the theory of post-translationally modified acetylated tau being a very early/upstream event in the pathogenesis of AD. We are currently conducting such a study at our center.
Laval University Research Center
Although correlative studies strongly support associations between hyperphosphorylated tau, tangles, and AD symptoms (Tremblay et al., 2007; Bennett et al., 2004; Ghoshal et al., 2002), investigations in animals have not yet confirmed how tau acquires its pathogenicity. Post-translational modifications, which may lead to conformational changes and aggregation, have been pointed out, including not only phosphorylation, but also glycosylation (Zhu et al., 2014) and acetylation (Min et al., 2015; Irwin et al., 2013; Kingwell, 2015).
In this nice paper, Tracy et al. follow up their group’s recent publication in Nature Neuroscience (Min et al., 2015) on the role of acetylation in tau pathogenicity, this time attempting to better define causal mechanisms using transgenic mice expressing human tau with lysine-to-glutamine mutations at K274 and K281 (tauKQ) to mimic acetylation. Interestingly, reductions in tau phosphorylated at Ser202/Thr205 and to a lesser degree at Ser396/Ser404 were observed in tauKQ mice. Their main finding, however, is that mimicking acetylation of tau at two sites impairs synaptic plasticity and memory retention, which complements well the observations made in AD brains. Indeed, tau acetylation was found to appear early in the disease, particularly for ac-K281, at least based on clinical dementia ratings and Braak scores. However, more detailed correlative studies with different tau phospho-epitopes retrieved in soluble/insoluble fractions and antemortem cognitive symptoms would probably help better define the role of tau acetylation in the disease.
Among mechanisms downstream of tau acetylation proposed by the authors, they identify a disruption of signaling pathways related to postsynaptic kidney/brain (KIBRA) protein which is well known for its role in synaptic plasticity and episodic memory, based on initial genetic studies (Milnik et al., 2012; Papassotiropoulos et al., 2006; Blanque et al., 2015). Incidentally, KIBRA modulators have been under development as cognitive enhancers by Sygnis Pharma for a few years (Pogacić Kramp and Herrling, 2009). A nice connection is also made with sirtuins, which are known for their deacetylase activity in vivo. A reduction of SIRT1 has been reported in AD brains, correlating with the accumulation of insoluble tau (Julien et al., 2009). Re-establishing SIRT1 activity could thus provide a means to correct the hyperacetylation of tau.
There is no question that this kind of study is the loom upon which we will ultimately weave our understanding of tauopathies. Whether acetylation is more pathogenic than other post-translational alterations and whether reducing tau acetylation at lysine 274 or 281 is a viable therapeutic strategy, however, remain to be determined.
References:
Tremblay C, Pilote M, Phivilay A, Emond V, Bennett DA, Calon F. Biochemical characterization of Abeta and tau pathologies in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2007 Dec;12(4):377-90. PubMed.
Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004 Mar;61(3):378-84. PubMed.
Ghoshal N, García-Sierra F, Wuu J, Leurgans S, Bennett DA, Berry RW, Binder LI. Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer's disease. Exp Neurol. 2002 Oct;177(2):475-93. PubMed.
Zhu Y, Shan X, Yuzwa SA, Vocadlo DJ. The emerging link between O-GlcNAc and Alzheimer disease. J Biol Chem. 2014 Dec 12;289(50):34472-81. Epub 2014 Oct 21 PubMed.
Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, Sohn PD, Schilling B, Cong X, Ellerby L, Gibson BW, Johnson J, Krogan N, Shamloo M, Gestwicki J, Masliah E, Verdin E, Gan L. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. 2015 Oct;21(10):1154-62. Epub 2015 Sep 21 PubMed.
Irwin DJ, Cohen TJ, Grossman M, Arnold SE, McCarty-Wood E, Van Deerlin VM, Lee VM, Trojanowski JQ. Acetylated tau neuropathology in sporadic and hereditary tauopathies. Am J Pathol. 2013 Aug;183(2):344-51. PubMed.
Kingwell K. Neurodegenerative disease: Targeting tau acetylation attenuates neurodegeneration. Nat Rev Drug Discov. 2015 Nov;14(11):748. PubMed.
Milnik A, Heck A, Vogler C, Heinze HJ, de Quervain DJ, Papassotiropoulos A. Association of KIBRA with episodic and working memory: a meta-analysis. Am J Med Genet B Neuropsychiatr Genet. 2012 Dec;159B(8):958-69. Epub 2012 Oct 12 PubMed.
Papassotiropoulos A, Stephan DA, Huentelman MJ, Hoerndli FJ, Craig DW, Pearson JV, Huynh KD, Brunner F, Corneveaux J, Osborne D, Wollmer MA, Aerni A, Coluccia D, Hänggi J, Mondadori CR, Buchmann A, Reiman EM, Caselli RJ, Henke K, de Quervain DJ. Common Kibra alleles are associated with human memory performance. Science. 2006 Oct 20;314(5798):475-8. PubMed.
Blanque A, Repetto D, Rohlmann A, Brockhaus J, Duning K, Pavenstädt H, Wolff I, Missler M. Deletion of KIBRA, protein expressed in kidney and brain, increases filopodial-like long dendritic spines in neocortical and hippocampal neurons in vivo and in vitro. Front Neuroanat. 2015;9:13. Epub 2015 Feb 20 PubMed.
Pogacić Kramp V, Herrling P. List of drugs in development for neurodegenerative diseases: update June 2009. Neurodegener Dis. 2009;6(4):165-212. PubMed.
Julien C, Tremblay C, Emond V, Lebbadi M, Salem N, Bennett DA, Calon F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009 Jan;68(1):48-58. PubMed.
SUNY Downstate Medical Center
The molecular mechanisms of long-term memory can be divided into two mechanistically distinct phases—induction and maintenance. Induction is a transient period of cellular consolidation lasting an hour or two, in which short-term memories are converted into long-term memories. Maintenance is the persistent phase of memory storage, lasting days to weeks to months and even longer. Which phase of memory does Alzheimer’s disease disrupt? Most neuroscientists would say induction. In part, this is because there are hundreds of molecules that have been implicated in induction, including NMDA receptor, CaMKII, PKA, ERK, mTOR, CREB, and the GluA1 subunit of the AMPA receptor, many of which are altered in the brains of individuals with Alzheimer’s disease or in animal models of the disorder. In contrast, only very few molecules have been implicated in maintaining long-term memory storage.
An important finding of Tracy et al. is that pathophysiologically altered tau, a hallmark of Alzheimer’s disease, might directly disrupt the persistence of memory through downregulation of one of these key maintenance molecules. A leading hypothesis for the molecular mechanism of long-term memory storage is the persistent action of the autonomously active PKC isoform, PKMζ, whose sustained phosphorylation maintains an increased number of GluA2 subunit-containing AMPARs at postsynaptic sites. The key molecule linking PKMζ to GluA2 is KIBRA, which directly binds both molecules.
Tracy et al. present data suggesting that in Alzheimer’s disease, abnormally acetylated tau disrupts the function of KIBRA, suppressing both long-term potentiation and long-term memory. Thus, the abnormally acetylated tau disconnects PKMζ, the biochemical mechanism of memory maintenance, from its target, the AMPARs that mediate functional modifications in neural circuits. If further work supports this hypothesis, this would mean that the tau-mediated pathology of Alzheimer’s strikes at the very essence of long-term memory storage.
Make a Comment
To make a comment you must login or register.