Rubbing Microglia the Right Way? At ADPD, Scientists Showcase New Strategies
Quick Links
Microglia seem to play a hand in every aspect of Alzheimer’s pathogenesis, from the seeding and clearing of plaques to the traveling of tau to the dying of neurons. Scientists are pursuing therapeutics that both promote their neuroprotective functions and put the kibosh on their neurotoxic behavior. At AD/PD, held March 5-9 in Lisbon, Portugal, researchers detailed preclinical findings on a handful of potentials coming down the pike. One was an antibody, discovered in the blood of cognitively sharp centenarians, that blocks the inhibitory receptor CD33, thus freeing microglia to carry out their protective duties with gusto. Other approaches, including an antibody and small molecule, activated TREM2, promoting microglial phagocytosis and plaque clearance. Finally, a new antibody reportedly mops up ASC specks, which are inflammatory, amyloid-seeding protein complexes unleashed upon activation of inflammasomes in microglia.
- An anti-CD33 antibody quells pro-inflammatory signaling in microglia.
- Antibodies and small molecules that activate TREM2 promote neuroprotective microglia, reduce plaques.
- Anti-ASC speck antibodies curb plaque seeding and gliosis.
Ralph Minter of London-based Alchemab Therapeutics presented preclinical findings on ATLX-1088, an antibody that blocks CD33 signaling. As an inhibitory receptor CD33 quashes phagocytosis and other microglial functions, so blocking it seems a promising strategy. However, Minter and colleagues did not set out to do just that. Rather, they started by hunting for autoantibodies tied to resilience to AD. That might sound a bit strange, because as Minter noted autoantibodies generally get a bad rap for causing a plethora of diseases, including multiple sclerosis, Type 1 diabetes, and rheumatoid arthritis, among others. However, in some cases, autoantibodies can protect against disease. For example, anti-HER2 antibodies protect against breast cancer. Likewise, the anti-amyloid antibody aducanumab was discovered in healthy older people who had little or no amyloid in their brains (Apr 2013 conference news).
To find more of these resilience antibodies, Minter and colleagues sequenced B cell receptors—the transmembrane precursors of antibodies—found in the blood of participants resilient to amyloid plaques in the European Prevention of AD consortium. Who was considered resilient? Any older person with blood biomarker evidence of Aβ accumulation but high scores on the MMSE and normal plasma p-tau. Minter said the goal was to find people who appeared stalled in the preclinical stage of AD, perhaps protected from progression. Antibodies to CD33 emerged as some of the few that were common among resilient people, but were not found among those with AD, Minter said. Next, Minter collaborated with Henne Holstege of Amsterdam University Medical Center to look for potentially protective antibodies in healthy centenarians in the 100-plus observational study (Oct 2022 conference news). Again, CD33 antibodies stood out. From this pool, the researchers selected their lead ATLX-1088.
In Lisbon, Minter reported that the antibody boosted phagocytosis of Aβ by human iPSC-derived microglia. In tri-cultures with neurons, astrocytes, and microglia, the antibody dampened the rise in multiple inflammatory cytokines in response to adding lipopolysaccharide and interferon-γ, suggesting it evokes a general anti-inflammatory effect, Minter said.
By mutating residues in CD33, the researchers narrowed down the epitope to CD33’s ligand-binding site, suggesting ATLX-1088 competes with the natural CD33 activator, sialoglycan. Other CD33 antibodies, including gemtuzumab, approved to treat acute myeloid leukemia, did not engage the ligand-binding site. Rather, several triggered internalization of CD33 by myeloid cells, effectively stripping the receptor from the cell surface and bringing the antibodies along for the ride. ATLX-1088, on the other hand, did not change levels of CD33 on the cell surface, functioning instead as a classic stoichiometric inhibitor of ligand binding. Minter said this bodes well for pharmacokinetics. Since CD33 is expressed on myeloid cells in the blood, any internalization would whittle down the amount that makes it to the brain. Ongoing experiments in mice expressing humanized CD33 in place of the mouse version support that idea, Minter reported.
How might naturally occurring CD33 antibodies confer resilience to amyloid? That is not known. Minter also does not know if these antibodies get into the brain in natural carriers, but for therapeutic development, Alchemab plans to target microglia.
What about other autoantibodies that might be floating around in healthy centenarian plasma? Michael Heneka at Luxembourg Centre for Systems Biomedicine wanted to know if TREM2 antibodies were among them. Minter said they have not found any, yet. He also noted that blocking a receptor, such as CD33, is generally more straightforward than activating one, as scientists are trying to do with TREM2.
Turning On TREM2
Indeed, scientists continue to move TREM2 agonist candidates toward the clinic, with Alector’s antibody, AL-002, furthest along, being evaluated in an ongoing Phase 2 trial in people with AD. Denali halted its DNL919 program last year after reports that the TREM2 antibody caused anemia; Novartis has a new candidate to fill the void. In Lisbon, Dominik Feuerbach from the company’s site in Basel, Switzerland, presented preclinical findings on VHB937. A fully human antibody, it latches onto TREM2’s ligand-binding, IgSF domain. Feuerbach reported that in iPSC-derived microglia and in cultured human M2A macrophages, VHB937 stabilized TREM2 expression on the cell surface, which boosted signaling through the receptor. This activation bolstered TREM2-related function, including phagocytosis of microglial “prey,” such as bacteria and apoptotic neurons, Feuerback reported.
In hTREM2-APP23-PS45 mice, which develop aggressive amyloidosis and express humanized TREM2, Feuerbach reported that administering weekly abdominal injections of VHB937 to 3-month-old mice resulted in fewer dystrophic neurites around Aβ plaques two months later. When the researchers injected these mice with a dye to label the plaques and then isolated microglia 24 hours later, they found more plaque material in the microglial innards if the mice had been treated with VHB937. This suggested that the antibody upped microglial appetite for plaques.
Boosting TREM2 signaling in this way also protected neurons in other disease models. In a PD model, the antibody spared neurons in the substantia nigra from the neurotoxin MPTP. It also accelerated remyelination in the wake of cuprizone treatment, a model for multiple sclerosis, Feuerbach reported. Collectively, the findings suggest that TREM2 agonism by VHB937 protects neurons from a range of insults, he said.
Vigil Neurosciences, based in Watertown, Massachusetts, has been developing small molecules to turn on TREM2 in people with AD. At last year’s AD/PD meeting, Vigil’s Christian Mirescu reported that the company’s top TREM2 agonist compounds appeared to work like “molecular glue,” by huddling TREM2 receptors together on the cell surface and boosting their signaling. In humanized TREM2 mice, and in nonhuman primates, Mirescu reported that the agonists crossed efficiently into the brain. Levels of the extracellular domain of TREM2, aka soluble TREM2, fell in the CSF, an indication that the agonist slowed processing and internalization of the receptor (Apr 2023 conference news).
The company has since selected a lead compound—VG-3927—as a candidate for AD trials. In Lisbon, Mirescu reported that when 4.5-month-old, plaque-ridden 5xFAD mice were fed with the agonist daily for six weeks, plaque area, as well as levels of insoluble Aβ42, fell by around 40 percent. The microglia adopted a neuroprotective, disease-associated microglia (DAM)-like signature, Mirescu said. Vigil has begun Phase 1 dosing studies with VG-3927 in healthy volunteers, with data due later this year.
One attendee asked Mirescu if he thought that chronic treatment with a TREM2 agonist might provoke tau pathology. This possibility was raised by findings from some mouse studies. For example, one study reported that activation of TREM2 with an antibody made tau pathology worse (Oct 2022 news). However, the relationship between TREM2 and tau is far from settled, as other mouse studies came to conflicting conclusions, or found that the relationship between TREM2 and tau may change as disease progresses (Oct 2017 news). Mirescu acknowledged these equivocal findings from mouse studies. To his mind, genetics from the tauopathy field speak for themselves, in that mutations that hobble TREM2 function raise disease risk (Guerreiro et al., 2013).
Shooting at Specks
In addition to targeting the dueling TREM2 and CD33 surface receptors, scientists are attempting to modulate microglia by way of inflammasomes. These multiprotein complexes contain nod-like receptors such as NLRP3, which serve as intracellular sentinels that detect danger signals, including Aβ and signs of cellular damage. Once triggered, inflammasomes set off a cascade that unleashes a storm of inflammatory cytokines that wreak havoc on neurons. Previously, studies led by Heneka implicated the NLRP3 inflammasome in AD, and various inhibitors are being developed to curb activation of the pathway (Dec 2012 news; Nov 2019 news; Li et al., 2023). In Lisbon, Heneka focused on apoptosis speck-like protein complexes, aka ASC specks, which are protein conglomerates that help form the inflammasome. They are released once microglia die a pyroptotic death. Heneka previously reported that ASC specks fuel Aβ aggregation in mouse models of amyloidosis, and that ASC proteins reside within the cores of Aβ plaques in the human brain (Dec 2017 news).
Heneka showed this aggregation frenzy happening in real time. Scientists in his lab generated transgenic APP/PS1 mice in which microglial CX3CR1 and ASC specks are fused to different fluorophores. Then, they injected methoxy-XO4 dye to label Aβ plaques, and monitored what unfolded in the brain with in vivo, live, two-photon imaging through cranial windows. Within the brain, they spotted microglia with ASC specks inside, as well as extracellular ASC specks that had presumably been released upon microglial death. Importantly, at the heart of every single blue, methoxy-labeled Aβ plaque, lay a ruby-red ASC speck.
To investigate whether the ASC specks were truly seeding plaques, the researchers checked the brain at different times after injecting the methoxy dye. They found that over the course of nine weeks, the amount of amyloid around each ASC speck grew, suggesting that ASCs had helped seed the plaques, which then proceeded to expand around the ASC core (image below).

Seeded by a Speck. In APP/PS1 mice injected with methoxy-XO4, ASC specks (pink) formed the core of plaques (blue) at baseline (left). Three (middle) and nine (right) weeks later, the ratio of plaque surface area to ASC speck volume increased. [Courtesy of Michael Heneka, Luxembourg Centre for Systems Biomedicine.]
Finally, Heneka reported that this toxic interaction could be thwarted with an antibody directed against the pyrin domain of ASC, which binds Aβ42. In an in vitro assay, the antibody blocked ASC-induced Aβ42 aggregation. Heneka would not disclose any information about the antibody or its development as a therapeutic.
Heneka does believe that ASC makes a good therapeutic target for AD, and that severing its ties with Aβ could thwart the seeding of Aβ plaques. While the inflammasome plays an important physiological role in fending off infections in the rest of the body, Heneka said that in the context of neurodegenerative brain, they do more harm than good. “I believe that you don’t need ASC specks in your brain,” he said.
Along those lines, Davide Basco from AC Immune, Switzerland, described preclinical findings from the company’s anti-ASC antibody, ACI-6635. Scientists at AC Immune used their SupraAntigen vaccine platform, which consists of liposomes embedded with antigens of choice, to evoke anti-ASC antibodies in mice. One, ACI-6635, bound to both mouse and human ASC with high affinity.
In Lisbon, Basco explained that ASC specks released from dying microglia not only go on to seed Aβ plaques, they can also be taken up by neighboring cells, where they propagate the inflammatory cascade. Nipping both processes in the bud is the goal of developing an anti-ASC antibody, he said. To see if the ACI-6635 antibody could break the inflammatory cascade of released specks, Basco treated cultured macrophages with human recombinant ASC aggregates to simulate release of specks. In response, the cells activated the inflammasome, and released IL-1β. Treatment with ACI-6635 potently blocked this response. It appears to work by promoting phagocytosis of the antibody-ASC complex via the antibody’s effector function.
Next, Basco tried the antibody in the ARTE10 transgenic mouse model of amyloidosis. It carries mutant human APP and PS1 genes. Starting at 3 months of age, when plaque growth was starting to ramp up, the mice were given 14 weekly injections of 60 mg/kg ACI-6635 or an isotype control antibody. In the controls, gliosis exploded, as gauged by levels of both Iba1 and GFAP detected in brain extracts. Immunostaining revealed diffuse ASC staining around Aβ plaques, surrounded by activated microglia and astrocytes. Basco emphasized that these diffuse deposits differ from the well-defined structures of specks. ACI-6635 reduced the amount of ASC protein in the vicinity of plaques, and reduced plaque size. The treatment also blocked the gliosis response almost entirely, as it dramatically stemmed the recruitment of microglia and astrocytes (image below).
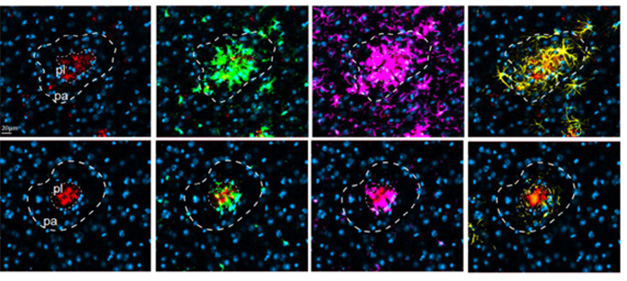
Calling Off Glia. In control ARTE10 mice (top), plaques cores (red) are surrounded by ASC (green), microglia (pink), and astrocytes (yellow). Treatment with ACI-6635 (bottom) lowers ASC, shrinks the size of plaques, and interrupts recruitment of glia. [Courtesy of Davide Basco, AC Immune.]
After the talk, Minter asked if the antibody’s Fc receptor, which was necessary for it to promote phagocytosis, might inadvertently provoke a damaging pro-inflammatory cascade, as has been seen with other active and passive immunotherapies. Basco said that had yet to be tested. Delphine Boche of the University of Southampton, U.K., wondered if Basco had tested for markers other than Iba1 and GFAP, to ensure that the antibody had calmed, but not killed, microglia and astrocytes. Basco said no, but he thinks the antibody works by changing the way microglia and astrocytes respond to plaques, and by keeping the cells in a healthier, less reactive state.—Jessica Shugart
References
Therapeutics Citations
News Citations
- Safe at 4 Grams? No ARIA at High Dose of Human Aβ Antibody
- Sharp at 100+? Thank You, Genes. Thank You, Immune System.
- All Roads Lead to TREM2: Gearing Up to Target This Receptor
- In Mice, TREM2 Antibody Mobilizes Microglia, Yet Worsens Tangles
- Changing With the Times: Disease Stage Alters TREM2 Effect on Tau
- Microglia and AD—Does the Inflammasome Drive Aβ Pathology?
- Microglia Inflammasome Stokes Tau Phosphorylation, Tangles
- Do Microglia Spread Aβ Plaques?
Research Models Citations
Paper Citations
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis Group. TREM2 variants in Alzheimer's disease. N Engl J Med. 2013 Jan 10;368(2):117-27. Epub 2012 Nov 14 PubMed.
- Li N, Zhang R, Tang M, Zhao M, Jiang X, Cai X, Ye N, Su K, Peng J, Zhang X, Wu W, Ye H. Recent Progress and Prospects of Small Molecules for NLRP3 Inflammasome Inhibition. J Med Chem. 2023 Nov 9;66(21):14447-14473. Epub 2023 Oct 25 PubMed.
Other Citations
Further Reading
Papers
- Harrison D, Billinton A, Bock MG, Doedens JR, Gabel CA, Holloway MK, Porter RA, Reader V, Scanlon J, Schooley K, Watt AP. Discovery of Clinical Candidate NT-0796, a Brain-Penetrant and Highly Potent NLRP3 Inflammasome Inhibitor for Neuroinflammatory Disorders. J Med Chem. 2023 Nov 9;66(21):14897-14911. Epub 2023 Oct 24 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.