At an expert meeting convened by Alzheimer’s Research UK, scientists from Eli Lilly shared Aβ biomarker data from the Expedition 3 trial with colleagues in academia and pharma. Reading the trial’s tea leaves together, the group felt that solanezumab entering the brain from the blood might have brought Aβ along for the ride, confounding CSF biomarker measurements. To boot, the trial appears to have enrolled participants with more brain amyloid than its predecessors. Given the weakness of the biomarker results, scientists, despite a day of deliberation, were left with merely a gut feeling that the hint of a cognitive benefit in the trial was probably real, if too small. A lively brainstorm session of where AD translational research should go next concluded the day. Read Gabrielle Strobel’s two-part story.
Solanezumab: Did Aβ ‘Reflux’ From Blood Confound Target Engagement in CSF?
In the aftermath of solanezumab’s failure to stem cognitive decline in mild Alzheimer’s disease, Eli Lilly and Company are closing down a Phase 3 trial in prodromal AD and laying off employeeswho had been hired in anticipation of bringing this anti-Aβ antibody to market. While Lilly retrenches, its senior scientists are also traveling to small gatherings around the country and abroad to share additional Phase 3 data with leaders in academia and industry. Their goal is to inform, and to learn how to prepare for better success next time.
A recent ARUK Roundtable took place near (no, not in) The Queens Larder. Located at Queen Square, London’s "Neurology Central," this pub takes its name from Queen Charlotte. She lived at the square and reportedly rented a cellar underneath the pub to store food for her husband, King George III, while he was being treated nearby for what is now thought to have been bipolar disease.
One such meeting was hosted by Britain’s AD research charity Alzheimer’s Research UK on January 19 in London. Lilly’s Eric Siemers disclosed results of solanezumab’s CSF biomarker measurements in the Expedition 3 trial, in addition to data shown previously at CTAD. Debating the data with fellow scientists and competitors at Biogen, Janssen, and Merck, Siemers, along with his colleague Mike Hutton, explored possible explanations for solanezumab’s disheartening performance. Also taking part in the conversation were Bart De Strooper, who leads the U.K.’s nascent Dementia Research Institute; Paul Whiting, John Skidmore, and John Davis of the U.K.’s tripartite Drug Discovery Institutes in London, Cambridge, and Oxford, respectively; as well as 10 academic leaders at universities throughout the United Kingdom, France, Canada, Sweden, and the United States. Research funders included Hilary Evans and colleagues from ARUK, Tetsuyuki Maruyama from the U.K.’s Dementia Discovery Fund, Rob Buckle from the country’s Medical Research Council, Martin Rossor from the National Institute of Health Research, among others. Mike Hobday of the World Dementia Council was there, as well as Elena Becker Barroso, who edits The Lancet Neurology.
Before the assembled group dug into the data, John Hardy of University College London applauded the openness to data-sharing and academic collaboration Lilly has practiced in its Alzheimer’s drug development. “We appreciate the frankness and the commitment, and we feel the genuine disappointment on the part of so many people involved,” Hardy said. He asked the group to come to a conclusion by the end of the day about whether the solanezumab program thus far had, in fact, tested the amyloid hypothesis. Hardy co-organized the roundtable with Evans and other leaders at ARUK. Lilly scientists will formally publish the Expedition 3 data; an expert discussion held in the meantime, to help the field at large learn in a speedy fashion, is summarized below.
First, a brief summary of this humanized antibody as presented by Siemers. Its clinical program dates back 13 years, to 2004, when Phase 1 data showed that solanezumab bound Aβ40 monomer in the plasma of AD patients much like the original mouse version, m266, had done in transgenic mice. Lilly also determined that 0.1 percent of injected solanezumab enters the CSF. The scientists took these two findings as indications that solanezumab would engage its target, and from there calculated that monthly injections of 400 mg of antibody would reduce free plasma Aβ by more than 90 percent. That became the Phase 3 dose, which corresponds to 5 to 7 mg/kg. Initially in Phase 2, Lilly was running a dosing arm of weekly 400 mg injections, i.e., four times higher and closer to what current antibody programs are using in 2016; however, the company ended it after deciding that the lower dose would max out the antibody’s efficacy. After all, the operative idea for solanezumab at the time was the peripheral sink hypothesis, which holds that drawing down free Aβ in blood would gradually reduce it in brain, as well, by way of dynamic equilibria connected across the blood-brain barrier.
It did not work out that way. In discussing why not, the ARUK meeting attendees kept returning to an observation from the early days of solanezumab research: When the plasma concentration of Aβ bound to solanezumab shot up as it did—about 8,000-fold after the antibody was infused—the half-life of Aβ shot up along with it. The half-life of free Aβ in plasma is on the order of hours, but that of the complex was 28 days. In other words, binding to solanezumab appears to have blocked the natural clearance pathway of Aβ.
The importance of this finding was unclear at the time. Moreover, in a subsequent Phase 2 trial, solanezumab treatment caused an increase in the CSF concentrations of total Aβ (i.e. bound and free), a decrease in free CSF Aβ1-40, and an increase in free CSF Aβ1-42. This led Lilly scientists to think that their antibody was in fact shifting the Aβ equilibrium in the brain somewhat. That interpretation won out over the fact that there was no evidence of plaque reduction, or cognitive efficacy, through Phase 2. The hope was that longer, larger trials would show both.
At the ARUK roundtable in London, scientists questioned whether free CSF Aβ42 should have gone up if solanezumab was working as expected. Some argued that any Aβ coming off plaques in response to a shifting equilibrium should have promptly stuck to antibody. Alas, in 2008, when Lilly was weighing the data it had, dose-dependent target engagement in blood, combined with Lilly’s active interest in the peripheral sink hypothesis, won out, and the company headed into Phase 3.
Fast-forward to January 2017, after three Phase 3 trials in some 4,100 patients. This news story will skip over the 33 percent slowing in cognitive decline in mild cases pooled from Expedition 1 and 2, and the cognitive and prior biomarker data in Expedition 3. These results were previously reported (Aug 2012 news; Dec 2016 conference news). What happened to CSF Aβ—i.e. target engagement, where the rubber hits the road—in Expedition 3? In London, Siemers reported that CSF total Aβ1-40 and Aβ1-42 rose in the treatment groups in Expedition 3, replicating the result of Expedition 1 and 2. In contrast, free Aβ1-40 and free Aβ1-42 in the CSF showed minor decreases, some not statistically significant, and some similar to the placebo group. To boot, the CSF free Aβ data did not consistently replicate results from Expedition 1 and 2. By and large, CSF free Aβ looked flat, contrasting with the movement seen in Phase 2.
What could this mean? At the ARUK roundtable, some scientists said that they would have expected a robust, 18-month-long therapeutic response to reduce CSF free Aβ. In discussion, academic and industry scientists honed in on the long half-life of solanezumab-bound plasma Aβ. Some recalled that more than 90 percent of peripheral solanezumab was bound to Aβ. To their minds, this implied that greater than 90 percent of the 0.1 percent of the infused solanezumab that made it into the brain might have carried Aβ from the periphery along with it. This could account for at least some of the total Aβ measured in the CSF.
“The biomarkers may have misled us here,” said Henrik Zetterberg of the University of Gothenburg and UCL. It is unclear whether the solanezumab-Aβ complex measured in CSF was formed in the brain. Dominic Walsh, of Boston’s Brigham and Women’s Hospital and UCL, said this matches preclinical data showing that m266 does not alter free Aβ in mouse brain either; rather, the antibody simply increases levels of Aβ bound to it (Mably et al., 2015). In toto, the CSF data in Expedition 3 provide no objective support for target engagement, agreed Zetterberg, adding, “The huge effects in plasma and the very minor effects in CSF make the CSF data uncertain.”
In London, further discussion elaborated on the equally unexpected florbetapir PET results, which were first presented at CTAD in San Diego. In Expedition 3, Lilly made a positive amyloid PET scan an inclusion criterion after concluding that many participants in Expedition 1 and 2 had likely been amyloid negative. This conclusion was extrapolated from a small amyloid PET sub-study done in those earlier trials, but it resonated with what had been seen in ADNI, AIBL, and indeed postmortem series. The decision to ascertain amyloid positivity in Expedition 3 was fueled by doubt in the accuracy of a clinical-only diagnosis as much as by growing enthusiasm for amyloid PET at the time. After all, Lilly had bought its own tracer, Avid’s florbetapir, and was in a leading position to use it for a large multicenter trial. Surely, weeding out amyloid-negative “non-ADs” would lift what was otherwise meant to be a confirmatory trial up above the small 33 percent effect size netted by the analysis of the pooled Expedition 1 and 2? Moreover, the Expedition 1 and 2 PET sub-study had suggested a statistically significant reduction in aggregated amyloid with solanezumab. Even though plaque reduction was not seen in mouse models, this first Phase 3 PET result was now fanning hope of a peripheral sink effect in people.
Again, it was not to be. In Expedition 3, plaque load nudged down just a smidgen, about 10 times less than seen with aducanumab. What happened? In London, Siemers explained that Expedition 3 participants had to have a florbetapir PET scan that was judged to be positive by way of a visual read. Researchers agree that visual reads tend to equate to a slightly higher amyloid burden than if amyloid positivity is determined quantitatively against a numerical threshold on the PET standard uptake volume ratio (SUVR). Indeed, in Expedition 3, the visual reads correspond to baseline SUVRs well above that threshold, Siemers said. Quite possibly, Expedition 1 and 2 may have included people whose amyloid burden was growing but was still below the SUVR threshold; they would have been screened out in Expedition 3. Site leaders for mild and prodromal-stage trials note that the cutoffs for biomarker requirements—which are still being refined as sponsors gain experience using them—sometimes force them to exclude participants who otherwise seem well-suited to a trial. “Our requirement for a visual read of the scan meant people had a relatively high amyloid load at baseline,” Siemers said.
This indication that neuropathology was more advanced in Expedition 3 than in Expedition 1 and 2 participants draws indirect support from the trial’s demographic data. They show that its groups were well-matched across categories, except that Expedition 3 enrolled about 67 percent ApoE4 carriers whereas Expedition 1 and Expedition 2 enrolled about 57 percent. ApoE4 is associated with amyloid deposition starting earlier in life. “When you require amyloid positivity in a trial, your ApoE4 carrier rate goes up,” Siemers said. In essence, changing the inclusion criteria meant the trial enrolled a different population, in this case slightly more advanced disease that may have been out of reach of the therapeutic oomph of solanezumab.
Overall, solanezumab had little effect on downstream markers of pathology or neurodegeneration. The CSF tau measurement showed so much heterogeneity among people that scientists at the ARUK roundtable considered them mostly noise. Commenting on the MRI findings, Nick Fox of UCL noted that the trial’s brain volume data showing no significant difference with solanezumab made sense. “In every study where there has been engagement with amyloid plaque, for example ARIA cases in immunotherapies, there has also been short-term increased brain volume loss. That we did not see this fits with amyloid PET not changing. We can conclude that solanezumab has not changed the established amyloid plaque load, with its associated inflammation,” Fox said.
Given those difficulties, was the 11 percent cognitive benefit reported in Expedition 3 true, or noise? The ARUK roundtable leaned toward the former view, because all cognitive curves across Expeditions 1, 2, and 3 separated in the same direction. Four of six cognitive readouts, each assessed by a different rater, showed a benefit. Siemers readily agreed that the 11 percent benefit was smaller than donepezil’s. “At Lilly, we agree that a benefit smaller than 15 percent is not clinically meaningful,” Siemers said. “It is not enough, and we cannot be sure it’s real.”
Did the 33 percent slowing of cognitive decline in Expedition 1 and 2, followed by 11 percent in trial 3, represent a “winner’s curse”? In other words, was it the regression toward the mean that often happens in GWAS and epidemiology, when the first test of a given sample nets a larger effect size than the replication study? Or was it a true biological effect of the 0.1 percent of infused antibody that reached the CNS, even if biomarker data does not support a clear mechanism of action? For his part, Hardy told Alzforum that given the measurement variability in clinical science, both Expedition1 and 2’s 33 percent and Expedition 3’s 11 percent cognition effect sizes could be within the 95 percent confidence interval for such studies. “It could be essentially the same result,” Hardy said. He compared solanezumab to the first airplane flight by the Wright brothers at Kitty Hawk in 1903. Their plane was in the air for all of 3.5 seconds and landed on its nose, yet the attempt was critical for success soon after.
The scientists assembled in London were curious about the dosing of solanezumab. Would it make sense to jack it up in the two remaining trials of solanezumab, DIAN-TU and A4? After all, a strong positive result in those two earlier-stage populations would be a tremendous boost for participants and the field at large. Some scientists cited dose increases in current trials of crenezumab and gantenerumab, though Biogen’s Samantha Budd Haeberlein argued against viewing anti-Aβ antibodies as a class. Each is sufficiently different in its binding characteristics and clearance mechanisms that they are no more similar to each other than one small-molecule drug is to the next. Others said that without a clear mechanistic rationale, they would not increase the dose mid-stream in a trial.
Lilly scientists explained that their early clinical studies were limited. For one, they never determined a maximally tolerated dose—a measure that is usually developed as a ceiling at which a drug becomes intolerable. In fact, in 2009, when Expedition 1 and 2 began, this international Phase 3 program of monthly antibody administration was straining Lilly’s manufacturing capacity. Manufacturing yields have risen since then, but at the time the constraint was real. Moreover, the saturation of plasma Aβ by 400 mg/kg of a biologic drug thought to work via peripheral sink made the dosing data seem adequate. In a recent Alzforum comment, Steve Paul, now at Voyager Therapeutics, noted the limitations of solanezumab dose finding. His point drew wholehearted agreement among scientists in London, who noted wryly that Paul was at Lilly at the time.
Be that as it may, industry and academic scientists in London strongly agreed that it appears the peripheral sink hypothesis of amyloid reduction has been refuted. Future antibody programs will approach dose-ranging differently, Siemers said. For example, Lilly and AstraZeneca are co-developing a new antibody to monomeric Aβ42 based on its CSF exposure and pharmacokinetics, not plasma.
Typically, after suffering a stinging loss, pharma companies consign the drug at hand to the scrap heap of history, pick another candidate from the pipeline, and soldier on developing that. But wait! Couldn’t much more use be made from at least a billion dollars’ worth of human research? After all, this program—which De Strooper could not help but mention cost multiple times the budget he gets to build a nationwide dementia research institute—could be argued to have conducted essentially a single experiment. Surely, more can be learned from its rich trove of samples and data.
At the ARUK meeting, scientists volunteered ideas and their collaboration. For one, Zetterberg noted that the CSF measurements thus far reveal but a sliver of Aβ biology in the brain. The major Aβ species in human AD brain is Aβ 4-42. It forms on plaques and can be measured after depleting Aβ 1-42 from the sample. “Looking at more species would be very important,” Zetterberg said.
The PET data warrant close dissection, as well. “This was the first time a global Phase 3 trial used PET as an inclusion criterion. It’s wonderful that there is so much PET data available in Expedition 3,” said Budd, to heads nodding around the room. Budd recommended Lilly drill down to individual-level data and compare people with higher or lower baseline amyloid, or indeed tau. Did they have lower cognitive scores within the baseline distribution? Did they progress faster? Did people respond differently to solanezumab based on their amyloid or tau burden at baseline? Did baseline amyloid or tau load rightly correlate with any other measures of the trial?
Luc Buee of the University of Lille, France, suggested stratifying Expedition 3 data in many more ways than shown thus far. “We’ve only seen ApoE4. We could stratify by other polymorphisms identified in GWAS, additional biomarkers including cytokines, chemokines, and other immunity/inflammation proteins, or even cell analyses such as macrophage, and T cell measures,” Buee said. “There is much going on in AD besides amyloid. We should learn about those things from a large trial like this to help us answer the question of whether solanezumab has completely failed, or too little of it got into the brain.”
About this question, De Strooper urged Lilly to conduct small exploratory studies pumping solanezumab directly into the brain’s intrathecal space. “Do not throw solanezumab away. It is a great tool. Put it directly into the brain, so you get much higher exposure. We know it is safe, we know it binds Aβ, we have a small hope it has an effect,” De Strooper said. Intrathecal infusion is considered impractical to deliver repeat doses to millions of patients, but it can be scientifically informative. It is, in fact, being used in clinical trials, for example in an international, multicenter Phase 1 study of an antisense oligonucleotide, aka ASO, to knock down huntingtin in people with early Huntington’s disease (see clinicaltrials.gov); preclinical research is showing promise for ASOs in tauopathies, as well (see Jan 2017 news).
The Expedition 3 discussion concluded with broad consensus around the room that solanezumab had not fully tested the amyloid hypothesis. Where next? “I worry that we keep making half-finished pointillist paintings: a dot here and a dot there, but we never see the picture,” said Hardy. He argued for systematic efforts to integrate more information into what is still an overly simplistic, amyloid-only view of Alzheimer’s disease. For more on that, see Part 2 of this series.—Gabrielle Strobel
After Solanezumab: Where Should Alzheimer’s Research Go?
At a postmortem discussion for solanezumab therapy in mild Alzheimer’s disease, held January 19 in London, members of the top echelons of AD research in the UK and other countries discussed CSF biomarker data from the Expedition 3 trial shared by scientists from Eli Lilly and company (Part 1 of this series). From there, they explored where the field should head next. Their agenda was not only to take stock of where the amyloid hypothesis stands after this latest clinical setback, but also to articulate research priorities for their meeting host, Alzheimer’s Research UK (ARUK) and for funders and the field at large. How should AD research evolve in 2017? While aducanumab, verubecestat, and other drugs are wending their way through Phase 3 (see Therapeutics database), how should translational researchers broaden their lines of attack?
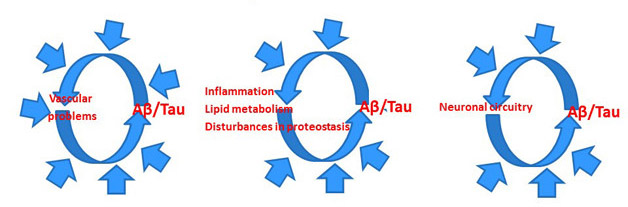
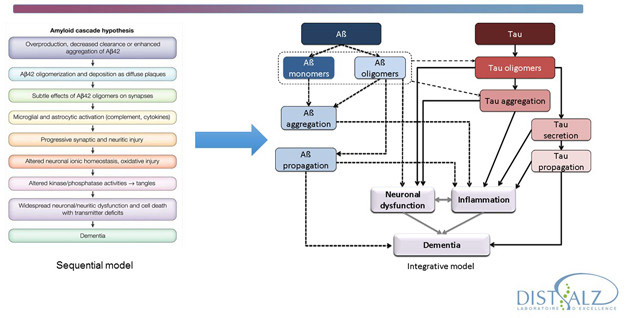
In short, the researchers agreed, they should do it by integrating more information into the amyloid hypothesis. Much more information, in fact. To be sure, solanezumab has not tested the amyloid hypothesis and current trials should continue. But at the same time, the hypothesis must change. In the 25 years since its inception, it has remained too rigid, said Bart De Strooper, Dementia Research Institute, London. Drug discovery still treats it as a linear concept, ignoring evidence that AD comprises parallel, and often circular, pathways of proteostasis, lipid metabolism, and glial activation. Each of these pathways interact with Aβ and tau in ways that remain poorly defined. “It’s not always that it starts with amyloid and ends with dementia,” De Strooper said, “Aβ is not equal to AD, so we need to think much more carefully.”
Rather than a linear amyloid hypothesis, researchers are conceptualizing circular, multicausal models for Alzheimer’s pathogenesis. [Courtesy of Bart De Strooper.]
The same goes for tau. “We really need to settle what is going on between the two main pathways. In trying to depict how tau relates to Aβ we draw lots of arrows, but we do not know what the arrows are,” said Luc Buee of University of Lille, INSERM, France. At the ARUK meeting, researchers in industry and academia agreed that the field in general has been too content to draw arrows between individual areas that are themselves somewhat understood, while leaving the arrows unexplained. The challenge now is to find ways to connect pathways to one another, because the whole makes up AD pathogenesis. “How do we link areas where we have some knowledge, and fill those mechanistic gaps?” is how Mike Hutton of Eli Lilly and Company put it.
Amyloid and tau pathways need to be integrated. Much of the research effort lies in understanding what mechanisms lie behind the links currently depicted as arrows. [Courtesy of Luc Buee.]
This work can start with GWAS hits and other genetic data. John Hardy of UCL noted a current example, whereby studying co-regulated variants in genes such as Trem2 and Abi3, as well as other genes including ABCA7 and even ApoE, is surfacing a theme of insufficient repair of damage to membrane lipids subsequent to amyloid formation. This kind of research requires a tremendous amount of work, De Strooper noted. It is worth the effort because it not only explains complex events downstream of amyloid formation but may also yield druggable targets. Finding new targets outside of Aβ remains essential, as a decade or more of clinical trials of non-amyloid therapeutic approaches has been every bit as disappointing as trials of amyloid-based drugs (see non-amyloid drug trials in Therapeutics ).
Pathways derived from new genetics need to be modeled in cells and animals, preferably supported in a systematic way as is being attempted in the U.S.-based MODEL-AD initiative (see Jan 2017 news; Dec 2016 conference news). Tau needs knock-in mice, Buee said. Beyond that, one new way of humanizing mouse models is to transplant AD-derived PSC neurons into mice and study their function and deterioration over time.
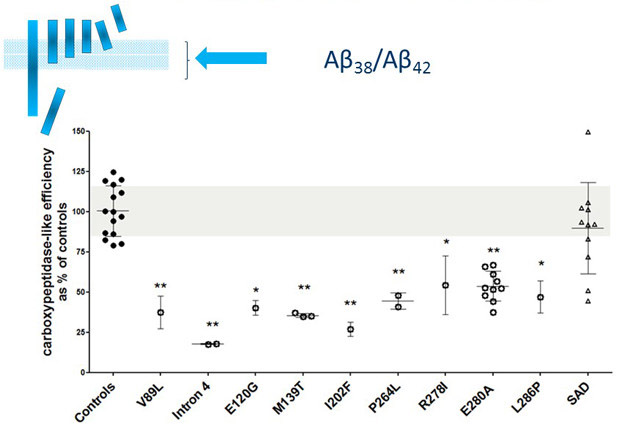
Drug discovery efforts targeting Aβ may consider using new measures that are informed by mechanistic research on how presenilin mutations make the γ-secretase carboxypeptidase function less efficient. [Courtesy of Lucia Chavez-Gutierrez.]
Separately, drug discovery research could make its measures more meaningful by incorporating advances about how presenilin mutations affect APP processing. For example, the Aβ40/42 ratio is a biologically arbitrary measure. “We measure it simply because we can,” De Strooper said. An Aβ38/42 ratio would better capture the current understanding that most mutations render the γ-secretase complex’s carboxypetidase activity inefficient.
To make oligomeric Aβ more tractable, the field should move beyond the stage where each lab has its favorite species. It could generate some consensus about the most important ones, and define those molecularly and functionally, De Strooper said. Other speakers at the ARUK meeting bemoaned how Alzheimer’s research tends to produce flashy papers and then move on to the next story. This leaves drug discovery to deal with lots of tantalizing but unsubstantiated ideas instead of producing converging evidence and tangible drug targets, said John Skidmore of ARUK’s Cambridge Drug Discovery Institute. For example, on the key question of how Aβ is neurotoxic, myriad papers proffer a list of potential therapeutic targets across receptor and membrane interactions. In practice, however, this literature suffers from "oversimplified thinking," De Strooper said, where one lab draws broad conclusions from a specific finding in a given experimental system, and too few labs replicate in a collective effort to weed out false starts and validate some central truths.
On toxicity, the sense at the ARUK roundtable was that Aβ does not directly kill neurons in AD. Why? Largely because toxicity is a slow process. The researchers agreed that structurally defining the most harmful forms of Aβ, which might only constitute a small fraction of the total, remains a priority. The path from those forms to neuronal death might be indirect, and go through tau or glia. On tau, the evidence that neurofibrillary pathology parallels regional neurodegeneration in AD is compelling. On glia, it is emerging. For example, a recent paper pegged three microglial cytokines as coaxing forth a neurotoxic subpopulation from among nurturing astrocytes (see Jan 2017 news).
Researchers emphasized that the term “neuroinflammation” is misleading in Alzheimer’s. “We should call it a different term,” said John Kemp of Janssen’s Neuroscience Discovery Europe.
Studies comparing gene expression modules show that Alzheimer’s does not cluster with bona fide inflammatory diseases, including multiple sclerosis. Likewise, CSF cytokine profiles indicate that what is going on in AD differs from what is classically thought of as neuroinflammation. Rather than framing their thinking in terms of neuroinflammation, AD researchers might be better served by delineating pathways of micro- and astrocytosis. AD-specific molecular phenotypes among glia might bear druggable targets on their surface.
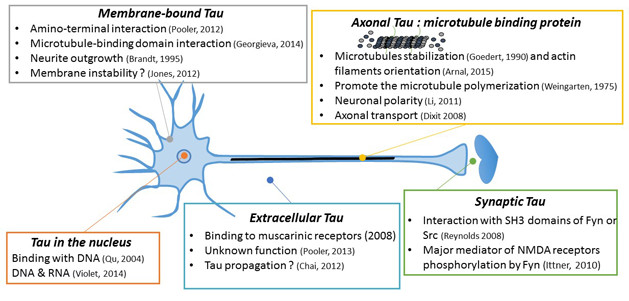
In theory, tau’s complex biology offers many angles for therapy development. [Courtesy of Luc Buee.]
As for tau, its gene is not a heavyweight in human AD genetics. Even so, the protein’s central role in Aβ neurotoxicity is undisputed thanks to research in neuropathology, CSF, disease staging, and cell and animal models. Moreover, tau PET is starting to link tangle pathology more tightly to cognitive decline than amyloid plaques. Being a microtubule-associated, but also a secreted and aggregating protein that is made in numerous splice variants, tau’s complex biology theoretically offers numerous flanks for attack by therapies. Alas, tau has been an elusive target.
Despite much effort in pharma companies and academic labs, no safe and effective drugs to inhibit tau’s hyperphosphorylation have been found. Few even made it to clinical trials. Indeed, biopsy studies have made clear that certain kinds of tau phosphorylation are physiological and lost postmortem (see Nov 2016 news). “We have to be very careful which phosphorylated tau we target therapeutically,” Buee said.
These different approaches to drug development for tauopathy are in different stages, from preclinical development to clinical trials. [Courtesy of Luc Buee.]
The first clinical trials of drugs targeting tau have failed (see Therapeutics database) and the Phase 3 aggregation inhibitor LMTM is widely seen to have been negative, as well (see Dec 2016 conference news). As of early 2017, tau immunotherapies are showing some promise in the clinic, though Buee cautioned that it’s early days, and characterization of their targets is limited (see Therapeutics).
As a field, Alzheimer’s research has produced fewer druggable targets than other disease areas, said Skidmore. That is true at least of classical targets such as enzymes and receptors. Take ApoE, or the high-profile area of protein propagation, which describes anatomical spread qualitatively but does not show toxicity, or generate handles for drug discovery. “We need to learn how to tackle classically un-druggable targets such as protein-protein interactions,” Skidmore said. Paul Whiting, of the ARUK Drug Discovery Institute at UCL, suggested that AD research advance from an era where it relied on pathologists and geneticists to a future of mechanistic and quantitative models. To this end, researchers urged AD funders to attract engineers, lipid biologists, and immunologists, and to fund them for a sufficiently long period so they can make their mark in a new field.
After years of research, the immediate causes of dementia remain mysterious, scientists conceded. Different levels of investigation, with their wildly different tools and languages, are each advancing on their own, but have not yet linked up a upstream cause—say, accumulated Aβ or tau—to the erosion of memory and function people experience as disease. Is it synaptotoxicity? Neuronal cell death? Glial activation? Circuit degradation? To solve this old question, scientists at the London roundtable argued for focusing resources on the convergent finding that it takes 15 years from amyloid to dementia, or indeed from a pretangle to a ghost tangle. This long period of enormous homeostasis of the brain should be characterized with systematic, genome-wide and cell-wide approaches. “I am dreaming of an atlas of AD, where we see how expression of twenty thousand genes in 100 different cell types evolves over time, Hardy said.
In addition, the group recommended to AD funders and leaders that they prioritize these areas:
Stay the course with approaches being taken at the moment. Science is learning a lot.
In parallel, support work that uses genetics to define new pathways.
To supporting pathway analysis, remove obstacles to public availability of genetic data.
Use genetics to strengthen clinical trials, e.g. by developing risk scores that boost the predictive value of ApoE and help identify at-risk participants for early stage trials.
Support ongoing clinical trials: speed recruitment, unify registries, improve retention.
Support development and validation of more sensitive readouts that can become surrogates for clinical benefit, e.g., wearable technologies, digital apps, home-based activity monitoring or cognitive testing. Better tools can help sponsors make better decisions before entering Phase 3.
Build biomarkers into trials across drugs and sponsors. Enable biomarker data from across trials to be analyzed together in such a way that the best markers are being iteratively validated for use by all sponsors. Foster the organizational cooperation needed to achieve that. In analogy, genetics started out with small, irreproducible signals, but generated meaningful results once large sample collections were pooled.
Improve education about translational research and clinical trials. Fund university courses that teach how to advance basic science to clinical trials. Educate journal editors to aid selection of rigorous clinical trial manuscripts; educate the public about the importance of trial participation. Foster contact between scientists and patients.
There is much to do to put therapy development for Alzheimer’s on a stronger footing, the roundtable agreed. Lest ideas drift apart, however, they concluded with a reminder of what unites them. Alzheimer’s research in 2017 suggests this broad umbrella is probably true: Everyone develops some tau pathology as they age; some people also develop amyloid pathology; in them, tau pathology spreads and prompts a microglial and astrocytic response; this leads to Alzheimer’s dementia. Vascular, metabolic, and other factors impinge. “Let’s not overcomplicate. This basic thinking frame remains useful,” Janssen’s Kemp concluded.—Gabrielle Strobel


Comments
No Available Comments
Make a Comment
To make a comment you must login or register.