Ironically, perhaps, progress in Alzheimer's diagnosis—think CSF and amyloid PET—may boost the profile of other dementing diseases that are often misdiagnosed as AD in clinical practice. This has not gone unnoticed among researchers on frontotemporal dementias (FTDs). They are combining their newfound ability to definitively eliminate AD as a cause of a patient's symptoms with significant advances in the science of FTDs themselves.
Out comes a public-private initiative to press ahead with therapeutic trials. In June 2012 in Washington, D.C., academic and industry leaders in FTD met with scientists at the Food and Drug Administration for a one-day powwow of what's in hand versus what still needs to be done in order to launch well-designed therapy studies for these emotionally wrenching neurodegenerative diseases. The need is desperate. No single drug is approved for FTD, and the many other drugs these patients receive barely help them, experts say.
Scientists Strategize With Regulators for Frontal Assault on FTD
Picture this: Your husband gets fired from his college professorship. He had written his students’ final essays himself and graded his work as theirs; they turned him in. Soon after, he buys a sports car he can ill afford and you beg his neurologist to get the dealer to take it back. Or this: Seized with chest pain, you drop onto the floor and urge your spouse to call 911. Unmoved, he replies, “Oh. What’s for dinner?” Such vignettes of executive and emotional dysfunction hint at why frontotemporal degeneration is a crushing disease, particularly for caregivers. They also animated a meeting that the FTD Treatment Study Group (FTSG) convened 4 June in Washington, D.C., where FTD clinician-researchers from North America and Europe met with experts from regulatory agencies, industry, family representatives, and public and private funders.
The questions of the day were, Given the explosive progress science has made in the past six years on the genetics and molecular pathology of this spectrum of dementing disorders, how close is the field today to running therapeutic trials that stand to be successful? If the field is not there yet, what is still missing? And what do scientists still have to do to attract the interest, funds, and expertise of the biotech and pharmaceutical industry?
Hosted jointly by the Association for Frontotemporal Degeneration (AFTD) and the National Institute of Neurologic Disorders and Stroke, the workshop drew 67 scientists from academia, industry, the U.S. Food and Drug Administration (FDA), and public and private funding organizations. It was the third major gathering of the FTSG since 2010, when the group started formalizing its work (see AFTD Web page). Previous meetings have laid out the general case for starting trials in these diseases, focusing specifically on preclinical studies (see ARF related news story) and clinical research (see ARF related news story).
In D.C., the academic scientists tried to persuade industry and regulators that they have largely put in place the pieces necessary to conduct therapeutic trials for these diseases. These pieces are the patients, the diagnosis, the molecular pathology, biomarkers, outcome measures. What, the researchers asked, do they now need to do to run trials that can take advantage of FTD’s orphan disease status and of the FDA’s existing mechanism of accelerated, aka conditional, approval? What advice can the agency offer to put clinical trials on a rational path early on?
“The goal for our meeting is to make a punch list of what needs to be done in the next year to accomplish successful FTD trials of disease-modifying agents,” said Adam Boxer of the University of California, San Francisco, who co-organized the day. To the industry scientists in the room, Boxer, who leads the FTSG, issued this challenge: “You should be developing drugs for this indication. You should be devoting significant resources. If that is not your opinion after today, we want to know what we need to do to convince you.”
By the end of a long day of talks and discussion, the scientists heard expressions of support from both regulators and industry for what they are trying to do. “You have impressed me tremendously. We are really interested in partnering with you,” said Lynne Yao of the orphan group with FDA’s Center for Drug Evaluation and Research. “This is a heroic effort,” said Michael Grundman of Global R&D Partners in San Diego, California. But at the same time, industry and regulatory scientists, speaking to the group and privately, made clear that the field still has to fill some gaps before pharma will jump in with its most promising drugs or the FDA will open the door to accelerated approval. The gaps include prodromal and drug response biomarkers, stronger consensus on outcome measures that can reliably pick up a drug effect amid the marked clinical heterogeneity of these diseases, and even fundamental questions of pathophysiology. “This is a wide-open, very young field,” commented one industry scientist. (This article does not name industry scientists to protect their ability to speak frankly at these meetings.)
Some of the answers will come from expanding ongoing natural history studies and from building international registries, particularly in genetic cohorts that follow presymptomatic cases over many years. Much as the Dominantly Inherited Alzheimer Network (DIAN; ARF related news story), the Alzheimer's Prevention Initiative (API; ARF news story), and the Alzheimer’s Disease Neuroimaging Initiative (ADNI) are doing for Alzheimer’s disease, and TRACK-HD is doing for Huntington’s (Stout et al. 2012), FTD registry studies could establish markers for disease progression and characterize the earliest features of FTD. Such studies can track how candidate fluid markers and imaging modalities change during the prodrome and symptomatic phase of disease, pinpoint when in life these various markers start changing, and clarify the relationship between important pathophysiologic proteins, for example, progranulin and TDP-43.
In particular, Lynne Yao from the Food and Drug Administration’s orphan drug group recommended that all stakeholders jointly fund an FTD registry study in support of all subsequent clinical trials, letting a patient organization manage it. “Drug development moves faster when patient-led organizations jointly push for a shared registry study that multiple sponsors can tap. Cystic fibrosis is an example where this is working,” said Yao.
Some of this is already underway. Howard Rosen of UCSF runs the Neuroimaging in Frontotemporal Dementia (NIFD) study on FTD and primary progressive aphasia (PPA); Boxer runs the 4 Repeat Tauopathy Neuroimaging Initiative (4RTNI) (4RTNI) on progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD); both are longitudinal and multicenter. A registry and biomarker study of genetic families and their asymptomatic carriers has been afoot in Europe for six months, with Martin Rossor and Jonathan Rohrer of University College London (UCL) at the helm, and government funds coming from the United Kingdom Medical Research Council and partners in other host countries. Called the GENetic FTD Initiative, or GenFi, it currently engages 13 centers in the UK, Canada, Italy, the Netherlands, and Sweden, with many more interested in joining, according to Jason Warren of UCL. “We … target recruitment of 30 subjects per centre, comprising 10 presymptomatic carriers, 10 mutation-negative family members, 10 affecteds. I believe this is the first consortium of its kind in genetic FTD and we are very excited about it,” Warren wrote to Alzforum in an e-mail. GenFi recruits across the spectrum of autosomal-dominant FTD, but targets the major genes MAPT (tau), PGRN (progranulin), and now C9ORF72. A genetic registry study is being planned in the U.S. While long and expensive, such cohort studies provide the foundation for drug development. “We spend a lot of time in my shop getting the natural history foundation for rare diseases. Once that is in place, the rest flows better,” Yao told the scientists.
What about clinical outcome measures, another key ingredient for successful therapy trials? They exist, but scientists agreed they need refining, ideally via a collaborative effort in the coming year. “Each of the measures needs work with the goal that we can achieve some consensus, hoping that, by the time trials start, we agree to design trials using similar outcome measures without going off in 10 directions at once,” said Howard Feldman of the University of British Columbia, Vancouver, Canada. On one candidate, magnetic resonance imaging (MRI), research across several independent longitudinal studies is beginning to produce convergent data by which atrophy corresponds to clinical function and tracks with disease progression. The brain also shrinks more rapidly with FTD than with AD. This was seen as a significant advance by academic and industry leaders alike, and suggests that MRI volumetry can cut down on the number of patients needed for clinical trials. At the same time, MRI volumetry for FTD is not fully standardized for multicenter trials. Other MRI modalities that show promise in FTD research, such as network connectivity imaging, remain experimental.
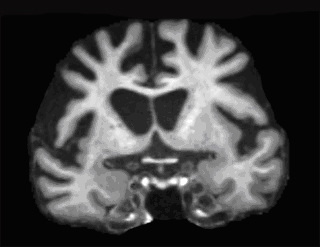
Coronal MRI scan through frontal lobes of a 59-year-old patient with behavioral variant FTD demonstrating frontal lobe atrophy. Image courtesy of Jason Warren
Fluid biomarkers for positive identification of FTLD are more limited, said Feldman. CSF tau in particular has proven obstreperous in FTLD. With current assays, this marker increases progressively in prodromal and frank AD, but curiously not in FTD, not even in "pure" tauopathies such as progressive supranuclear palsy or the genetic form, frontotemporal degeneration with parkinsonism-17 (FTDP-17).
The most definitive blood-based markers in FTLD are the genetic ones, for example, progranulin or, more recently, C9ORF72, both of which are linked to TDP-43 pathology. They allow researchers to identify the molecular pathogenesis underlying a given patient’s clinical disease. Deterministic gene mutations account for a larger fraction of FTD than do APP and the presenilin genes in AD; they may supply enough patients to power small trials, especially if several specialized sites band together. The monkey wrench for this scenario is, at present, that people with progranulin mutations can develop different phenotypic disease. Scientists still need to work out how these can all be aggregated into a single therapeutic trial using a single outcome measure. In essence, fluid markers at present seem able to sort populations for inclusion, whereas MRI and cognition/function might serve to measure drug effects (for more on imaging and fluid markers, see Part 2).
As is usually the case in planning meetings on pre-competitive issues, where multiple pharma or biotech companies are in one room, discussions steered carefully clear of candidate drugs. Representatives from 14 companies were present, but none mentioned even drug classes, much less individual "assets." This is as close as things came to naming anything: In response to a plaintive audience quip at the end of a long day, “Where are the drugs? Were they discussed while I was in the bathroom?” Boxer allowed this much: “Most companies in this space work on monoclonal antibodies against tau. I would like to see those go into progressive supranuclear palsy or an FTD indication before they go into AD.”
Frontotemporal degeneration has gained both prominence and financial support in recent years. The New York Sunday Times in May 2012 ran a front page feature in its Vanishing Mind series. Years earlier, the tauopathy progressive supranuclear palsy (PSP) penetrated public consciousness when the entertainer Dudley Moore spoke openly about coping with PSP in interviews with Barbara Walters and others.
Private funds for therapy efforts on the progranulin forms of FTLD have come from an affected family through the Bluefield Project. “We are looking for the best and brightest ideas to invest in this field while insisting on a highly collaborative environment,” Roy Richards, who co-sponsored the D.C. meeting, told the audience. Similarly, research on the tau side of FTLD has been boosted by the Consortium to Study Tauopathies at UCSF. Separately, the Tau Consortium is an initiative by the investment billionaire Richard Rainwater, who has PSP. Besides supporting this meeting, this consortium also funds the development of new research models, including iPS cell lines derived from cells donated by Rainwater. The meeting host, the Association for Frontotemporal Degeneration itself, included an RFA form in the conference materials as well. For more on this conference, see Part 2 of this series.—Gabrielle Strobel.
Toward FTD Therapeutic Trials: Diagnosis Firm, Outcomes Still Soft
On 4 June in Washington, D.C., scientists from academia, industry, and funding organizations met to discuss precompetitive steps in the preparation for clinical trials for frontotemporal degeneration. Frontotemporal degeneration (FTD) encompasses a group of diseases that are distinct from Alzheimer’s, the most common neurodegenerative disease. At the same time, FTD exists on a broad spectrum with AD, and a partial overlap in symptoms frequently leads to misdiagnosis of one for the other. Part 2 of this series summarizes components of future trials where clinician-researchers have reached broad consensus—that is, its subtypes and diagnosis—as well as others where more work is needed—that is, endpoints to measure the success or failure of a drug.
Frontotemporal degeneration was first recognized by the Czech neurologist and psychiatrist Arnold Pick, who described it as early as 1892 as a focal neurodegenerative disorder. “That was incredibly modern at the time,” said Bruce Miller of University of California, San Francisco. In 1957, French scientists published a detailed comparison of FTLD and AD (Delay et al., 1957; discussed in Thibodeau and Miller, 2012). Today, scientists of the FTD Treatment Study Group (FTSG) see FTLD as falling into three major groups. The behavioral variant (bvFTD) is a social disorder that starts with psychiatric symptoms, whereas two language variants called progressive aphasia are marked either by difficulties with semantic understanding (svPPA) or by fluency and grammar (nfvPPA/agrammatic PPA). FTLD is rare enough to qualify for orphan disease status (see more in Part 4 of this series), but is actually as common as AD among people age 45 to 64, when FTD strikes primarily.
A large fraction of FTLD cases are diseases of tau. This includes rare genetic forms such as FTDP-17, as well as sporadic conditions that have overlapping symptoms with movement disorders, i.e., progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD). More recently, tauopathies broadly have further come to include an anticipated wave of cases of chronic traumatic encephalopathy (CTE) generated by concussive or blast injuries in contact sports and war (see ARF related news story). “I think we will experience an epidemic related to tau,” Miller told the audience in D.C.
Other large fractions of the FTD spectrum are accounted for by the C9ORF72 and progranulin genes and by TDP-43 pathology; a small slice is attributable to the FUS and CHMP2B genes. For details on the current classification of FTD, see image.
In daily medical practice, particularly in primary care, FTD and AD can easily be misdiagnosed. Typically, people get the better-known AD diagnosis when, in fact, they have a form of FTD. While both diseases impair a person’s executive function, FTD erodes a person’s social and interpersonal behavior but spares memory and visuospatial function, which deteriorate in AD. Importantly, both FTD and AD now have new consensus sets of diagnostic criteria that emphasize the value of biomarkers, said Bradford Dickerson of Massachusetts General Hospital, who runs the largest FTLD research program in New England. At the AFTD conference, representatives of the Food and Drug Administration signaled that they see no problem with using these criteria for grouping patients in clinical trials (see Part 4 of this series). With this, one obstacle for good trials—sorting the right patients into the right trials and treatment groups—appears surmountable.
The subtypes of FTLD are sufficiently well understood to begin grouping them for trials by drug type, Dickerson said. For example, tau-based drugs could be tested in bvFTD, agrammatic PPA, PSP, and CBD, while people with semantic PPA or the motor neuron disease variant of FTD should be excluded. These last two forms of FTLD would be candidates for trials of drugs targeting progranulin or TDP-43.
For bvFTD, consensus clinical criteria (Raskovsky et al., 2011), together with a finding of frontal lobe atrophy on MRI or perhaps a negative amyloid PET scan, can render a diagnosis with great confidence, said Dickerson. The same is true for FTD’s language variants. There, new consensus diagnostic criteria describe the characteristics of the three clinical subtypes of PPA (Gorno-Tempini et al., 2011), and structural MRI reveals a stereotypical pattern of asymmetric frontoinsular atrophy that corresponds to the language symptoms. In a collaboration between Dickerson and Marsel Mesulam at Northwestern University, Chicago, atrophy patterns were consistent between patients seen at MGH and Northwestern. Longitudinally, too, independent MRI studies at different centers are generating data on atrophy rates that agree with each other and begin to support sample size calculations for trials, said Howard Rosen of UCSF. Overall, atrophy rates are nearly twice as fast in FTD as in AD, said Rosen.
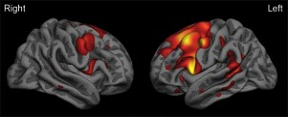
Surface map of the cerebral cortex from a group of patients with the agrammatic variant of primary progressive aphasia demonstrating left-greater-than-right frontal lobe atrophy (red-orange-yellow indicate areas of thinner cortex in patients versus similarly aged controls). Image courtesy of Jason Warren
Surface map of the cerebral cortex from a group of patients with the semantic variant of primary progressive aphasia demonstrating left-greater-than-right anterior temporal lobe atrophy (red-orange-yellow indicate areas of thinner cortex in patients versus similarly aged controls). Image courtesy of Jason Warren
Still, biomarkers are necessary to reduce diagnostic errors in multicenter trials, said Murray Grossman of the University of Pennsylvania in Philadelphia. On the imaging front, tau PET is desirable, as it specifies an underlying molecular pathology, which FDG-PET and MRI cannot do. However, PET is only just entering the first human studies (see 2012 ARF HAI conference report and Zhang et al., 2012), whereas the last two are already widely available. With PET, one concern beyond cost and radiation exposure is that it requires longer scanning times. Some FTLD patients simply do not lie still long enough to conduct a PET scan plus the MRI scan necessary for co-registration, scientists at cautioned in D.C. Partly for this reason, functional connectivity MRI is rising as an experimental modality in the FTLD field. For its part, structural MRI will soon become more specific with multimodal imaging combining gray and white matter, Grossman said.
All told, the general consensus of the day was that structural MRI, while not perfect, is advanced enough for use in therapeutic studies. “The clinical and imaging measures work for trials,” said Dickerson (see Sapolsky et al., 2010). Initial learning trials will precede subsequent registration trials, industry scientists cautioned.
While FTLD research lags behind AD research in establishing positive biomarkers for diagnosis, it benefits nonetheless from those successes in AD. In particular, amyloid PET scans will be a boon for FTLD trials, researchers agreed. “With amyloid imaging we can now predict with almost 100 percent certainty who is on an FTLD path and who is on an AD path. This is helping us tremendously to diagnose FTLD,” said Miller. Dickerson put it this way: “The approval of amyloid PET is going to revolutionize clinical trials. According to the FDA, the main indication is in identifying people who are negative for amyloid. That will expose more FTLD cases.” Ironically, perhaps, a milestone in AD research will raise the profile of other insidious diseases that have long languished in its shadow.
Progress is less clear on fluid measures. In blood, genetic markers for some forms of FTLD are both sensitive and specific. With 30 percent of cases having a family history, blood tests can capture a fair number of patients. The progranulin gene alone is thought to account for up to 10 percent of all cases of FTD. At the same time, the majority of cases are sporadic, and neither genetics nor proteomic studies have yet provided robust fluid markers for them. “We need large-scale proteomic markers,” said Grossman. CSF AD research is helpful in the same indirect way as amyloid imaging, in that the Alzheimer’s Aβ42/tau signature distinguishes FTLD from AD. However, CSF tau at present is curiously uninformative for tau forms of FTLD. In those, even in pure tauopathies, patients have the same levels as controls, perhaps because the assay does not capture the particular tau species altered in those diseases. A robust TDP-43 assay that works reliably in multiple centers is not yet available, and data on CSF progranulin remain sparse.
Once patients are enrolled into a therapeutic study, how will the trial measure success or failure of the drug? The day saw extensive discussion, but little consensus, about the quality of outcome measures in FTLD. In general, outcome measures for FTLD trials were seen as less solid than diagnostic criteria for inclusion/exclusion. While FTLD strikes younger, otherwise healthy people and progresses faster than AD, it is also more clinically heterogeneous, and this poses striking challenges for measuring outcomes. But the issue is solvable, said Howard Feldman of the University of British Columbia, Vancouver, Canada. “We struggle with how we look for drug effect, but it occurs to me that if people fall apart over one to two years, we should be able to capture that.”
To do that, different groups use different clinical rating scales. Some adapt tools for AD, such as multi-domain cognitive composites co-developed by David Knopman of the Mayo Clinic in Rochester, Minnesota, or an FTLD-specific version of the Clinical Dementia Rating Scale (CDRS), an established scale that measures behavior and everyday function. Both form part of an FTLD module recently added to National Alzheimer’s Coordinating Center (NACC) assessments (see ARF related news story). Other groups use their own ratings, such as the Addenbrooke’s Cognitive Examination-revised (ACE-R.) In short, the different domains affected in the different variants of FTD—cognitive, executive, psychiatric, global—have been examined with different tests and partially validated in different longitudinal multicenter studies. For measuring activities of daily living, the Functional Activities Questionnaire (FAQ) seemed acceptable to the assembled scientists. No clear frontrunner for use in treatment trials has emerged; however, the ACE-R has published reports of longitudinal sensitivity as well as translation into major world languages other than English to its credit. The standard 6-domain CDR has also been translated, but the newer FTLD-specific CRD as-yet has not.
The group discussed whether the Frontotemporal Temporal Dementia Study Group (FTSG) should develop its own "perfect" scales or use existing scales. Petra Kaufman of the NINDS offered this advice: “Do not make the perfect the enemy of the good. There can be huge effort in improving an outcome measure, and then, in the end, a very simple measure is eventually chosen.” This is true especially for international studies.
There was agreement that, in general, cognitive/functional scales may work better in trials than behavioral/psychiatric ones. For the neuropsychiatric inventory (NPI), in particular, scientists cautioned that patients falsely appear to improve on it as their disease progresses toward increasing apathy. They do "better" on the scale because they withdraw socially and commit fewer of the inappropriate acts (e.g., touching people, reckless spending) that the scale also captures. Essentially, the NPI captures positive and negative symptoms that can cancel each other out. Such scales are sensitive over the short term to measure a bump in symptomatic improvement, but likely not in long trials needed to show disease modification.
In FTD “long,” as in “needed for Phase 3,” will mean 12 months, not the 18 or even 24 months customary in AD, Knopman suggested. Likewise, Phase 2 trials ought to last, at the utmost, six months. That is because the disease progresses so quickly that many participants won’t stay within the dynamic range of assessments, or even be able to perform study tasks much past one year. When participants leave a trial prematurely, it loses statistical power.
What does all this amount to for therapeutic studies? “We must be honest that with the outcome measures we have, we will need fairly large trials. Because of FTD’s rarity, this may mean international trials; hence, the outcome measures will be administered in local languages and should be simple. But trials are definitely feasible,” Knopman said.—Gabrielle Strobel.
Case Studies Crystallize Trial Ideas at FTD Conference
Earlier this month, researchers and regulators took up an invitation from the Association for Frontotemporal Degeneration and the National Institute of Neurologic Diseases and Stroke to meet in Washington, D.C., for a day of work toward therapeutic trials for these diseases. After discussing the general requirements for FTD clinical trials (see Part 2 of this series), they got specific. To that end, they broke the heterogeneity of clinical syndromes and molecular pathologies of the FTLD spectrum of diseases into three case studies. Progressive supranuclear palsy (PSP) represents a sporadic pure tauopathy; progranulin-deficient FTLD represents a genetically defined entity, and behavioral variant FTD represents a clinical syndrome. Each stands for a patient population for whom the Frontotemporal Dementia Treatment Study Group (FTSG) would like to design therapeutic trials. In each case, an FTSG scientist presented a design and then solicited advice from regulators of the Food and Drug Administration and industry scientists.
Case Study 1: Progressive Supranuclear Palsy Michael Gold of Allon Therapeutics, Inc., in Vancouver, Canada, noted that as a pure tauopathy, PSP would seem an ideal disease for evaluating tau-based drugs free of the complicating influences of mixed amyloid and vascular pathologies that are often found in Alzheimer’s. PSP is the second most common parkinsonian disorder after Parkinson’s itself. Its core phenotype is recognized, its diagnosis pathologically validated, its clinical progression well known and inclusion criteria widely accepted. PSP worsens so quickly, with up to 20 percent annual mortality, that trials can be substantially shorter than trials in AD or PD.
The medical need is great, said Irene Litvan, a movement disorders specialist at the University of California, San Diego. Litvan said that almost none of the long list of medications her PSP patients tend to be on when they are referred to her actually help them. This includes antidepressants, antipsychotics, and cholinesterase inhibitors. The ALS drug riluzole has been tested in the largest PSP study to date, the Neuroprotection and Natural History in Parkinson’s Plus Syndromes (NNIPPS) trial; it did not help patients but was generally regarded as a well-conducted study that taught the field how to run PSP trials. The approved MAO inhibitor rasagiline is currently completing a one-year Phase 3 trial. The experimental GSK-3 inhibitor NP031112/tideglusib by the Spanish biotech company Noscira completed a Phase 2 trial, called Tauros, in late 2011. In December 2011, the Spanish news site elEconomista.es reported that it failed to achieve its objectives.
In D.C., Gold spoke about Allon’s ongoing Phase 2/3 trial of davunetide, a peptide delivered by intranasal spray. Following on the NNIPPS trial, the davunetide study confirmed that PSP trials are doable. Screening and enrollment occurred apace, and site investigators and raters are generating reliable data. “The logistics and feasibility for PSP studies are absolutely there,” Gold said.
Alas, Allon’s experience also highlights the challenges of evaluating drugs in PSP. It is difficult to find early-stage patients because many physicians do not recognize that patients have a neurologic problem and, hence, only refer them once the disease is well established. There are no criteria for prodromal PSP or validated biomarkers to improve diagnosis, response to treatment, or progression. Of the few choices for outcome measures in PSP, none has yet been validated in a therapeutic trial. The Movement Disorders Society is about to publish a critique of all available PSP scales. One specific problem Allon discovered was that, as the characteristic gaze disturbance of PSP worsens, some patients lose the ability to read. “Be careful with tests that require reading,” Gold said. What’s more, the rapid progression is a double-edged sword. On the one hand, it may reduce the number of subjects needed in a clinical trial as well as the length of the study. However, rapid progression may lead to more patients becoming unable to travel to their sites for repeat visits. Falls, in particular, can be dangerous, even fatal, because PSP patients appear to lose their ability to break a fall, Gold said.
In the Allon trial, about 20 percent of patients consented to lumbar puncture, Gold said. This low level of participation can be addressed in future studies. In other studies, including DIAN, ADNI2, and the Bristol-Myers Squibb prodromal AD trials, lumbar puncture consent rates approach 100 percent. What gives? Scientists agreed that the difference lies in how lumbar puncture is presented and explained to both site physicians and participants. “In ADNI2, we get even second lumbar punctures without a problem. Clearly, the first one was not so onerous that patients do one but refuse another,” said Murray Grossman of the University of Pennsylvania, Philadelphia. William Hu of Emory University, Atlanta, Georgia, added, “When I moved to Atlanta, our lumbar puncture rate went from 10 percent to 90 percent. It is how you explain it. You say this is a part of the study and it is important, and patients will participate. It takes a culture change.”
The Allon trial has set the stage for subsequent PSP trials to learn from its woes. “We have a mechanism that will make the data available to the research community. Even if our study did not work, it will put foundational pieces in place for the next one,” Gold said. Gold called for a global PSP registry for tracking progression in patients that multiple companies could tap to evaluate their compounds.
Regulators recommended that in the longer term, researchers develop a therapeutic trial strategy where individual drugs are first proven to have a pharmacological effect and are then tested together in adaptive combination trials, as pioneered by the I-SPY breast cancer study. “I believe it is very difficult to affect a neurodegenerative process with a single drug. Having a real clinical impact is probably a matter of a combination therapy, like in cancer,” said Christina Sampaio, a movement disorders specialist who in 2011 left the European Medicines Agency after 13 years to join the Cure Huntington's Disease Initiative, which funds and shapes therapeutic research in that disease. Sampaio spoke personally, not on behalf of the EMA. Industry scientists emphasized the need for pharmacodynamic biomarkers in PSP.
Case Study 2: Symptomatic Progranulin Deficiency Adam Boxer of the University of California, San Francisco, argued the case for trials to boost levels of the growth factor progranulin, whose genetically induced reduction leads to FTLD. No such trials have been done yet. “Correcting progranulin deficiency is the low-hanging fruit in neurodegenerative disease treatment,” said Boxer.
Progranulin is the second most common FTLD gene after C9ORF72, and 4 percent of sporadic FTLD patients have low levels of progranulin as well, said Boxer. The growth factor’s role in inflammation indicates it might be widely useful in neurodegenerative diseases that have an inflammatory component.
Toward this goal, the field has generated a range of mouse models of progranulin deficiency. Some existing drugs raise progranulin levels in mice (e.g., Cenik et al., 2011), and several companies are working on their own compounds that penetrate the blood-brain barrier and are safe enough for long-term therapy.
“Why are we excited about progranulin as a target? Because we can measure it in blood and think its elevation can become a surrogate endpoint in trials,” Boxer told the audience. CSF data indicate that progranulin levels of mutation carriers and non-carriers are clearly separated—a promising sign for clinical studies. Boxer peppered regulators and industry scientists with questions on what they would want to see done before granting an IND. What level of animal or iPS cell data is necessary to move a compound into humans?
Boxer started a conversation on trial design by proposing a dose-finding/proof-of-concept trial that would enroll progranulin mutation carriers. It would measure safety, tolerability, and serum and CSF progranulin levels with ascending doses of the compound in weekly increments until a maximum tolerated dose was found. Boxer asked industry and regulatory scientists these questions: What if we see a change in plasma but not CSF? Do we need to think about genes that modify progranulin expression? Does the trial need to show an increase of CSF progranulin, or would measuring blood levels suffice? Could serum progranulin be a basis for conditional approval? How large an increase in progranulin is enough?
This proposal generated ample discussion. Dana Hilt from EnVivo Pharmaceuticals in Watertown, Massachusetts, noted that moving progranulin levels up to the bottom of the normal range may be sufficient, since heterozygotes and non-carriers do not overlap. To do that, researchers must know the test-retest variability of the assay, as well as the variability of a given patient over a period of days and weeks. Regarding plasma versus CSF, Hilt recommended that the trial sample both, making plasma the primary endpoint and studying the latter. “In this early stage, I would explore dose ranging and various imaging outcomes in a learning study, which would help design a larger, more confirmatory study,” Hilt said.
Steve Salloway of Brown University, Providence, Rhode Island, advised the FTSG to stay agnostic at this early stage of progranulin research. “I know we have a desperate need to treat. But we are in new territory. Let’s gather information and be careful, not hasty,” Salloway said. Initial drug studies should show that the drug moves the biomarker. “The idea of moving from that point to conditional approval without showing a clinical benefit seems premature to me,” Salloway said. Extending natural history research to gain as much data on the disease as possible will pay off later, Salloway said.
Lynne Yao from the FDA’s Center for Drug Evaluation and Research agreed, noting that because every patient in these rare diseases is so precious, every study, even the first dose-finding study, should extend those patients and keep observing them to gather longitudinal data. “With rare diseases especially, all data will be important in the end. We look at the entire package from all studies, so make the most use of every patient,” she said.
Yao further noted that the scientists would have to show if serum and CSF progranulin correlate and explain how raising progranulin in plasma would make things better for the patient. If indeed it turns out that progranulin does not change in CSF in response to the drug, then plasma progranulin would be a diagnostic marker for inclusion, but not a surrogate, Yao said.
Several scientists recommended that progranulin models be used to explain the biology between progranulin and disease, and to test the assumption that progranulin itself is indeed the best step in the pathway to tackle therapeutically. Industry scientists agreed that more understanding of CSF progranulin and its downstream connections to TDP43 and disease would have to be in hand before they would launch large-scale trials.
Case Study 3: Behavioral variant FTD
What about symptomatic therapies? Treatments based on tau or progranulin would target a core molecular pathophysiology of FTLD and, hence, presumably modify disease. Conceptually, disease modification beats symptomatic treatment, and it dominates conversation at conferences. But proving disease modification of a progressive disease sets a higher technical bar than showing a temporary symptomatic benefit. Symptomatic treatments may be valuable, particularly for sporadic cases of FTD when no molecular cause is known.
In behavioral variant FTD, there have been two small trials of a symptomatic treatment that may provide a starting point for future work, said Elizabeth Finger of the University of Western Ontario, London. The neuropeptide oxytocin is a maternal hormone that is thought to promote social cognition in animals and humans (Donaldson and Young, 2008). It is made in the hypothalamus in neurons that project to the frontal lobes. Conceivably, oxytocin might help with the interpersonal deficits that make bvFTD emotionally wrenching, such as a patient’s inability to process facial expressions and recognize vocal affect in loved ones.
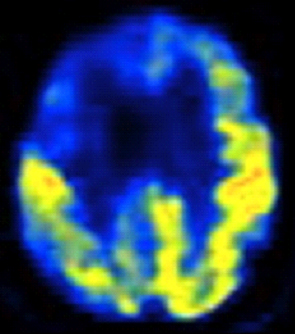
Axial FDG-PET scan through frontal lobes of a 59-year-old patient with behavioral variant FTD demonstrating frontal lobe (right greater than left) hypometabolism (right side of brain is on left side of image). Image courtesy of Jason Warren
Oxytocin can be given intranasally (Chang et al., 2012). Some human studies see a trend toward improved social recognition and emotional empathy (e.g., De Dreu, 2012). With that, Finger and colleagues ran a proof-of-concept study of a single dose of oxytocin spray, in which 20 people with bvFTD completed various tasks testing their ability to recognize facial and auditory expression and theory of mind (Jesso et al., 2011). This initial study recorded an improvement in the Neuropsychiatric Inventory (NPI) Questionnaire. Subsequently, Finger’s group tested three doses given to people with bvFTD for a week, with the same measures taken at one week and adverse events measured at two weeks. This generated no serious side effects, and the lower dose registered a small effect on apathy measures on the NPI. Responders inquired somewhat more about relatives and showed an interest in reading and daily activities, Finger said.
Given those data, Finger asked the audience if a multicenter study on oxytocin would be worth doing. In fact, she asked, are symptomatic treatments worth the cost and effort at all? In subsequent panel and audience discussion, this latest question generated a resounding “Yes.” Symptomatic drugs are clinically valuable, said Ilan Irony of the Food and Drug Administration, speaking for many. This is seen as true, particularly in the interim years until enough disease-modifying drugs are available to max out site, investigator, and funding capacity. “I might not go after symptomatic drugs if there were better alternatives, but right now there are not,” said Steve Whitaker from the biopharmaceutical company Omeros. With regard to moving forward with oxytocin, the consensus was weaker. Before tackling a larger trial, dose finding could be revisited with a study geared explicitly toward bvFTD, not based on cancer or other indications.—Gabrielle Strobel.
Take It From the Agency: First FTD Drug May Be an Orphan
One purpose of the meeting of the Frontotemporal Degeneration Study Group (FTSG) held 4 June 2012 in Washington, D.C., was to give its scientists an opportunity to show the Food and Drug Administration how far their research has come (see Part 1, Part 2, and Part 3 of this series). The other was to get feedback on how to proceed to ensure that future trials meet success. The conversation unfolded amid the cross currents of palpable excitement about the fundamental scientific advances in FTD versus a sobering realization that the pharmaceutical industry is shrinking back from neurodegenerative indications in the aftermath of negative trials in Alzheimer’s. “The exuberance about FTD drug development is justified. Let’s keep it rational,” said James Kupiec of Pfizer’s Neuroscience Research Unit in Groton, Connecticut.
How is the FTSG to do that? Step 1 is to consult early and often with regulators on both sides of the Atlantic, said Lynne Yao of the FDA’s Center for Drug Evaluation and Research (CDER). This Alzforum story first summarizes Yao’s general remarks to the assembled group of academic, industry, and funding scientists; Part 5 excerpts agency replies to FTD-specific questions the FTSG had submitted prior to 4 June.
First off, treatments for FTD fall under the auspices of the Orphan Drug Act of 1983, because this group of diseases affects fewer than 200,000 patients in the U.S. Similar legislation is on the books in Europe as well. The law is known for its financial incentives; however, equally important is that it provides drug developers more frequent access to free advice from regulators, and Yao urged the FTSG to take full advantage of this expertise.
For context, a third of CDER’s approvals between 2006 and 2010 went to orphan products. This includes both small-molecule drugs and biologics such as antibodies. One-fifth of rare disease approvals went to first-in-disease indications. Those programs tend to be unconventional, Yao said. CDER has allowed the use of historical controls, which are frowned upon in trials for common diseases. CDER has considered evidence in related populations, non-traditional study designs, and pharmacodynamic endpoints. Those approvals went to diseases affecting 150,000 to 180,000 patients. “Studies in very rare diseases are possible and can lead to approval,” said Yao.
One example is a genetic kidney disease called NAGS deficiency. In 2009, the European Medicines Agency (EMA) approved the drug CarbaGlu to treat it. The regulators did so based on only 12 patients in the dosed group, said Christina Sampaio, formerly of the EMA. In 2010, the FDA followed suit. What clinched the case? A package of 17 years' worth of data from fewer than 100 patients altogether, a blood biomarker as a surrogate outcome, and additional data from the literature. “This program used extremely out-of-the-box thinking,” Yao said.
So, if orphan drug status is possible, then why is it still tough to obtain? The answer is that the Orphan Drug Act does not provide a separate (read "lower") standard. “You still need the same evidence for effectiveness and safety,” said Yao.
In practice, the challenge of gathering that evidence with few patients available means that an orphan drug program typically starts out without qualified endpoints, outcome measures, or biomarkers, much less a surrogate endpoint. It’s learn-as-you-go, and datasets to establish those tools and then use them in registration studies will be small. For patients, the stakes are high because their rapidly progressing, fatal disease may leave but one shot at a trial. Yao put it this way: “For a disease with a huge number of patients, let’s say hypertension, if you pick the wrong endpoint and design a fancy study, you may get a mess of data and have wasted time and money, but you can enroll thousands more patients for a better second study. You do not have that luxury here. So I recommend that you plan carefully, because these patients desperately need you to get it right the first time.”
What does planning carefully mean? The clinical development plan should rest on natural history studies that generate information on pathophysiology and the mechanism of action of a planned intervention. That forms the basis of IND-enabling data, which open the door to clinical trials. In developing a coherent plan, the FTSG should bring everyone to the table, notably toxicologists, pharmacologists, and patient community representatives, Yao said. For this work in particular, the orphan designation offers ample access to regulators.
An IND then requires animal pharmacology and toxicology data to permit an assessment of whether the drug is reasonably safe, as well as data about manufacturing, product composition, stability, appropriate production, assurance that the drug will be available, and other information.
INDs for rare diseases vary, but invariably they open the door to the next big step: dose finding. This is difficult in rare diseases, Yao acknowledged, as more doses studied mean the groups become awfully small. Even so, it's critical. “You must do some dose finding. You can’t tell the dose based on a mouse, or a different human indication. We will not insist on five doses having been fully tested. But do tell us what dose makes sense based on good pharmacokinetics from your first human data,” Yao said.
Both the FDA and EMA have two ways of approving a drug. Besides regular or full approval, there is also accelerated approval, which came into being as a result of HIV/AIDS activism. It is granted when the drug moves a surrogate endpoint in a way that is reasonably likely to produce a clinical benefit later.
Researchers in the FTSG, and also AD researchers preparing Dominantly Inherited Alzheimer Network (DIAN) and Alzheimer's Prevention Initiative (API) trials, covet this. It sets a lower bar than showing clinical or even global benefits in prodromal, asymptomatic, or very clinically heterogeneous populations. But what does “reasonably likely to produce a clinical benefit” mean, exactly? There is no blanket answer, Yao said. Basically, the drug must come with substantial evidence of safety and likelihood to predict clinical effectiveness. That means the study clearly distinguishes the effect of the drug from other influences, for example, by using adequate controls and ensuring that the outcome instruments do not have their floor or ceiling in the study population and duration. Specifically, FTD markers that were discussed on 4 June—above all, serum progranulin—at present are far from amounting to a surrogate endpoint, Yao said. “But when you think you have one, talk to us early.”
Adaptive and Bayesian trial methods drew attention at the meeting, but Yao cautioned that in many rare diseases, too little is known about pathophysiology and the drug’s mechanism of action to execute such designs. Even though the orphan drug group at the FDA is sympathetic to adaptive approaches, in reality it sees few such trials.
With accelerated approval, the sponsor must conduct post-marketing research to show that the surrogate outcome indeed generates a clinical benefit later. When that fails, the FDA withdraws approval. Earlier this year, for example, the FDA revoked the metastatic breast cancer indication for Avastin, which the agency had conditionally granted in 2008.
Regulators at the EMA approach orphan drug development in much the same way as does the FDA. In particular, EMA scientists facilitate development planning, and orphan drug evidence must be as strong as for any drug, said Sampaio, who spoke personally, not on behalf of EMA, which she left in 2011 to head drug development for the Cure Huntington’s Disease Initiative in Princeton, New Jersey.
One difference between the FDA and the EMA concerns the terminology around surrogate markers. Thomas Fleming, a noted biostatistician at the University of Washington, Seattle, wrote an influential article in Health Affairs about the pitfalls of surrogate markers and accelerated approval (Fleming, 2005). Fleming’s classical definition requires proof that the effect of the drug on the surrogate is correlated with the effect of the drug on the clinical outcome, and that the size of the effects is correlated. “There is no surrogate in existence that fulfills these conditions. They are nearly impossible to prove,” Sampaio said. For this reason, both agencies, in essence, are moving away from the classic concept of the surrogate endpoint and toward the concept of cumulative evidence that the endpoint is connected to the clinical outcome. “This cumulative endpoint is a matter of judgment. It is a matter of acceptance by the scientific community. It is a matter of building history. It may take years or even decades,” Sampaio said. The terms "surrogate endpoint" and "accelerated approval" persist because they are written into FDA regulations, but for its part, the EMA has come to use the terms "cumulative evidence" and "conditional approval" instead, Sampaio said.—Gabrielle Strobel.
For FTD Drug Development, a Q&A With Regulators
In advance of a 4 June meeting in Washington, D.C., with regulators and industry scientists, Adam Boxer of the University of California, San Francisco, and other steering committee members from the Frontotemporal Degeneration Study Group submitted questions to the U.S. Food and Drug Administration (FDA). FDA scientists including Russell Katz of the Center for Drug Evaluation and Research (CDER) and Nicholas Kozauer from the center’s Degeneration Group huddled over those questions. As they were unable to attend the meeting, Sampaio agreed to present their answers, adding her own view in places. FTD clinical trial pioneers, here’s the redux:
Q: Compounds have entered clinical trials for the treatment of progressive supranuclear palsy (PSP) predicated on the notion that PSP represents a fairly homogeneous tauopathy. What is your view regarding the generalization of clinical trials from PSP to other sporadic tauopathies such as corticobasal degeneration (CBD), non-fluent/agrammatic variant primary progressive aphasia (nfvPPA) and behavioral variant FTD (bvFTD)? Do you need randomized clinical trials in other tauopathies, or would you consider biomarker confirmation of a drug effect on tau?
A: Tauopathy as a unified indication across clinical disorders is problematic unless you have a single outcome measure for them. At least one separate study would likely be required for each disease. The relationship between pathology and clinical presentations in these diseases is still emerging. One study in a new indication may be sufficient if a widely accepted relationship with PSP exists. This all presupposes that a biomarker for tau exists.
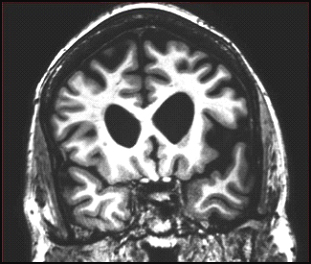
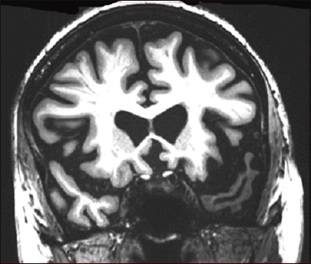
Coronal MRI scan through frontal lobes of a 62-year-old patient with the agrammatic variant of primary progressive aphasia demonstrating frontal lobe atrophy (on the left side of the brain, right side of the image). Image courtesy of Jason Warren
Coronal MRI scan through frontal lobes of a 66-year-old patient with the semantic variant of primary progressive aphasia demonstrating temporal lobe atrophy (on the left side of the brain, right side of the image). Image courtesy of Jason Warren
Q: Given the clinical and pathological heterogeneity of other sporadic tauopathies, would you recommend the use of enrichment strategies in trials, for example, a biomarker to identify pathology as a determinant for inclusion in the study?
A: Absolutely.
Q: Does the agency believe a non-specific label for the treatment of sporadic tauopathies is possible, analogous to neuropathic pain?
A: Neuropathic pain is a syndrome; its pathology is unknown. Ultimately, this is the surrogate question. You would have to demonstrate an understanding of the actions of the drug and the given disease.
Sampaio: The FDA does not give umbrella indications for brain syndromes; the EMA may be more likely to do that. It is too early to make the case that when we treat tau, we treat disease. Personally, I think that is where we are headed.
Q: Do you have a view regarding the current diagnostic criteria for the sporadic tauopathies?
A: They appear reasonable from a regulatory perspective.
Q: Do you agree that the diagnoses of PSP, FTD, and CBD are sufficiently well established to represent treatment indications for new therapies?
A: Yes.
Q: Regarding clinical trials in CBD, bvFTD, or nfvPPA, do you have a particular view on specific versus global outcome measures relevant to the diseases being studied? What about the need for co-primary endpoints?
A: Expect similar guiding principles as in symptomatic AD.
Sampaio: I personally take a different view. The CGI performs poorly in long trials. First of all, "CGI" stands for clinical global impression. In a long-term trial of a neurodegenerative disease, you do not expect improvement, so even the title of the scale is wrong. Secondly, the investigator is anchored to the baseline that can be 18 months earlier, so this, in effect, becomes a memory test for the rater. I think the FTSG should define innovative designs and create the data to support your proposals. You are the experts. Bring it to the agency; don't fight among yourselves, and then persuade us with a good argument.
Q: Do you have a view on what the clinically relevant benefit would be in these conditions?
A: No. That is why we require a global or functional co-primary endpoint.
Q: Do you have a view on the degree to which tau-directed therapies need to affect some or all isoforms, 3R versus 4R?
A: No. We are concerned with clinical outcome versus specified mechanism of action. We cannot comment on the acceptability of an effect on a given tau isoform as a surrogate at this point.
Q: Regarding translation of findings from PSP studies to Alzheimer’s patients, would positive PSP studies preclude the need for dose ranging in AD trials?
A: No. A relationship cannot be assumed.
Q: Would positive PSP studies provide supportive safety data for AD studies and reduce the need for long-term exposure in AD patients?
A: Yes.
Q: Would your view regarding the value of CSF tau/p-tau as a validated biomarker in AD change if treatment for PSP affected CSF tau/p-tau?
A: No. Again, the relationship cannot be assumed.
Q: With regard to developing a treatment for FTD in patients with progranulin mutations, how large a sample is needed to meet regulatory needs for safety?
A: There is no pre-specified exposure requirement for safety data in orphan conditions. We can be flexible.
Q: What outcomes might be convincing in presymptomatic carriers? Would raising serum progranulin suffice for conditional approval? If not, would a drug-placebo difference on a single composite measure relevant to disease be sufficient?
A: This is, again, the surrogate question. It requires a very detailed understanding of the MOA of both drug and disease. What would be the composite measure in presymptomatic disease? We are open to considering an argument, as in early stages of AD, where the impetus is toward specific cognitive outcomes, not global clinical ones.
Q: Preliminary work exists on clinical trial measures for FTD or tau populations. Some tools, such as the NPI, have been used extensively in AD trials as secondary outcomes. The CDER has been expanded to include domains relevant to FTD for use in FTD trials. How much validation would be required to accept those as an outcome measure for a clinical trial?
A: Face validity may be reasonable in otherwise accepted scales.
All told, the regulatory experts sent the FTSG on its way with these closing comments: “We want products for sufferers of rare diseases that actually work, so we want to base our decision on the best science. We are excited to work with any and all of you to figure out the most efficient path. Fast is not always most efficient,” said Yao. Sampaio concurred. She closed the day with a last point that industry had brought up in side conversations, as well. “People get excited about biomarkers. But remember that biomarkers are not interchangeable until you prove the relationships between them,” Sampaio said. “I see speakers make lists of imaging modalities and fluid outcomes, and then assign group sizes to each. Doing that is a recipe for failure. This is not a supermarket where you can shop the one that gives you fewer patients in the trial. Each marker measures a different thing, and you must understand how they relate.”—Gabrielle Strobel.

{kind=link}
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.