CONFERENCE COVERAGE SERIES
Human Amyloid Imaging 2011
Miami, FL, U.S.A.
14 – 15 January 2011
CONFERENCE COVERAGE SERIES
Miami, FL, U.S.A.
14 – 15 January 2011
The location alone left nothing to be desired, at least for snow-weary New England attendees who only the day before had been digging out from under 24 inches of white. But much more than palm leaves swaying in the breeze along a sandy beach distinguished the 5th Human Amyloid Imaging (HAI) conference, co-organized by Keith Johnson of Massachusetts General Hospital in Boston. Notably, a lineup of female speakers pushed the research envelope into the new and fiendishly complex terrain of multimodal and connectivity imaging. Their goal: Begin to get a grip on understanding how amyloid in the brain intersects with age, ApoE, cognitive reserve, glucose use, and many other factors in a person’s long slide toward Alzheimer’s disease.
Held in Miami Beach on 14-15 January 2011, this HAI conference for the first time unfolded as an independent meeting, having been linked to the American Academy of Neurology’s annual conference before. The organizers briefly wondered if HAI alone would draw people in a field where conferences big and small are growing in number. They need not have worried; the meeting filled to capacity, late registrants found themselves on a waitlist, and the program for the first time expanded to easily fill a second day.

At HAI, 18 of the meeting’s 47 speakers and session chairs were women. They spanned the scientific career ladder, from graduate students (e.g., Wenzhu Bi of the University of Pittsburgh Medical School and Elizabeth Mormino at the University of California Berkeley) to postdoctoral fellows (e.g., Kristen Kennedy of the University of Texas University in Dallas), staff scientists (e.g., Jessica Langbaum at the Banner Alzheimer’s Institute in Phoenix, Arizona, and Susan Landau of Berkeley), physicians (e.g., Noora Scheinin at the Turku PET Centre, Finland); from assistant professors (e.g., Prashanthi Vemuri of the Mayo Clinic in Rochester, Minnesota), professors (e.g., Julie Price of the University of Pittsburgh Medical School), senior PIs (e.g., Agneta Nordberg of the Karolinska Institute in Stockholm, Sweden, and Susan Resnick, National Institute on Aging), and a company CEO (Dawn Matthews of Abiant Inc.). Discussion panels followed each session, and one such panel featured the unusual sight of a lone man (Michael Devous from University of Texas at Dallas) seated next to five formidable women.
Sponsored by unrestricted educational grants from the usual suspects—that is, the companies that develop amyloid imaging agents and some pharma companies that make AD drugs—HAI awarded a Young Investigator prize and 11 travel fellowships to young scientists in this rapidly growing field. Patrizia Vannini of Brigham and Women’s Hospital, Boston, nabbed the former for her talk on how a brain area involved in both the default-mode network and episodic memory loses its agility when amyloid conspires with age (see Part 2 of this series). The latter went to Vemuri, Scheinin, Kennedy, Langbaum, Annie Cohen of UPitt; Stefan Foerster of the Technical University Munich, Germany; Jaana Koivunen of the Turku PET Centre; Dong Young Lee of Seoul National University College of Medicine; Karen Rodrigue of UT Dallas; Liang Wang at MGH; and Guofan Xu at the University of Wisconsin, Madison.
The program in Miami ran the gamut from fresh-off-the-bench data on exploratory mechanistic studies to a discussion with clinical leaders of how far the field has moved toward consensus on the best use of amyloid imaging in clinical trials. This arc from basic to therapeutic research was punctuated by studies that image astrocytes together with amyloid, one finding a surprise effect of taking antidepressant medication on brain amyloid, and an attempt to pin down just what microbleeds might have to do with amyloid deposition.
Perhaps the biggest change in this field in the past year has been that a growing number of research labs are branching out from amyloid PET-only studies and are now using PET as but one tool to probe what’s going on in the brains of both normally aging people and those who are on the road to AD. The finding, now reproduced several times, that a large fraction of aging people have fibrillar amyloid deposits in their brains has raised pressing questions about which other factors determine how well, and for how long, the brain can withstand its damaging effects. Scientists believe that multipronged research, ideally in the same people over time, will answer these questions. This research combines amyloid PET with FDG-PET to get a sense of how neurons fare metabolically when amyloid is around, with increasingly challenging cognitive tests to pin down subtle deficits, and with various paradigms of functional MRI to probe brain networks. The goal is to understand how the brain works, how its function relates to amyloid buildup, and to tease apart how aging interacts with genetics, brain reserve, and vascular disease.
On the industry front, company scientists and academic researchers contracted to test their ligands continued to present, as they did last year, studies they view as steppingstones toward regulatory approval. The HAI conference unfolded amid intense anticipation of an upcoming FDA Advisory Committee meeting. It was to give a thumbs up or down on a New Drug Application (NDA) for clinical application of the farthest advanced among a small handful of commercial amyloid PET tracers currently being developed, i.e., Eli Lilly’s AV-45/florbetapir, which was recently renamed Amyvid. (That meeting has since rejected approval of the NDA as is, but recommended eventual approval for Amyvid provided the sponsor meets further stipulations; see ARF related news story.) For more on HAI, see Part 2, Part 3, Part 4, Part 5, Part 6, and Part 7.—Gabrielle Strobel.
This is Part 1 of a seven-part series. See also Part 2, Part 3, Part 4, Part 5, Part 6, and Part 7. View a PDF of the entire series.
At the 5th annual Human Amyloid Imaging meeting, held 14-15 January 2011 in Miami, Florida, the bulk of the talks and posters reflected scientists’ grappling with how amyloid deposition in the brain behaves over time and what it does to the brain’s ability to function. The field appears to agree that it’s bad for the brain; beyond that, exploratory results and new ideas dominated the discussion.
Annie Cohen of the University of Pittsburgh Medical School, Pennsylvania, started things off with her new data on the fundamental question of how the amount of Aβ people harbor in their brains changes over time. The general idea these days is that brain amyloid begins to rise with age, in people’s fifties and sixties, about a decade earlier in people with ApoE4 than those without (and earlier still in people with autosomal-dominant mutations). This starts some 15 years prior to a clinical diagnosis of dementia; after that diagnosis, amyloid levels stay largely the same during the person’s last decade or so of life with mild to moderate, and then severe dementia. Biomarker staging diagrams shown at conferences these days (e.g., Jack et al., 2009; ARF Webinar) depict brain amyloid levels swinging up with age in an S-shaped curve, though in truth, researchers readily acknowledge they don’t know for sure yet what shape that curve takes, and whether it levels off completely. Some studies have suggested that the increases over time are more straight than S-shaped; others that brain Aβ keeps rising through the course of diagnosed AD dementia. “Whether amyloid deposition goes up linearly or in a sigmoid fashion is very important later for the use of biomarkers in therapeutic trials,” said Keith Johnson of the Massachusetts General Hospital. A definitive answer will come from more data on more people from more time points in longitudinal studies; meanwhile, here is the latest cut from Miami.
Cohen reported that in her group of 55 cognitively normal controls, 27 people with MCI, and 16 with AD, PIB retention over the course of three years did increase in more than half of those with AD. In the control and MCI cases, brain amyloid progressively rose in those who had started out PIB positive, and in some who had started out PIB negative, though many of the latter at this point remain below a set threshold of "PIB positivity." “We think amyloid accumulates in a linear manner over time,” Cohen said. In a study that primarily probed technical aspects of this question by comparing different imaging processing methods in ADNI participants, Susan Landau at University of California, Berkeley, also saw a small increase in amyloid load over time in people with AD.
In contrast, Jaana Koivunen of the PET Centre in the Finnish city of Turku reported data more in synch with the idea that PIB deposition is largely complete by the time of an AD dementia diagnosis. Koivunen follows 29 people in their seventies with MCI using PIB-PET, MRI, and neuropsychometry. She reported that 17 volunteers who met criteria for probable AD two years after baseline had started out with higher amyloid deposition at baseline than did volunteers who did not progress clinically, but these 17 added no further amyloid since baseline. In contrast, the 12 who clinically progressed little in those two years were still adding more amyloid, raising the question of whether they might reach the AD diagnosis in the future. Hippocampal atrophy increased in both groups, similar to earlier one-year follow-up data from the Mayo Clinic in Rochester, Minnesota (Jack et al., 2009). On a poster, Rik Ossenkoppele at the VU Medical Center in Amsterdam, The Netherlands, similarly reported that the curve rose in the MCI group and flattened off in AD.
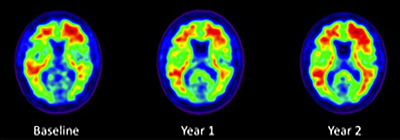
Three PIB-PET scans taken two years apart in an ADNI normal control show modest expansion of brain amyloid over the course of two years. Image credit: Susan M. Landau, UC Berkeley
In discussion, Michael Pontecorvo from Avid Radiopharmaceuticals noted that even his company’s larger dataset of many hundreds of patients imaged prospectively with florbetapir does not to date support the drawing of a more precise staging curve, as no one has yet figured out how to analyze all the factors that affect amyloid deposition over time. One problem, said PIB co-developer Chet Mathis of the University of Pittsburgh Medical School is that group averages obscure change in individual people. In a given person, scientists do see a large jump in one year, and a leveling off the next. “We will get at the shape of the curve once we have enough data on enough subjects for statistically sound modeling, but at this point we lose the dynamic view because we present group averages,” Mathis said. Scientists need more data to get a handle on a related issue, as well: namely the time course of when different brain areas, especially those affected later, deposit AD. These areas keep changing deeper into the course of disease, but no comprehensive view of their sequence in time is yet in sight.
Diagnosed AD aside, the earliest phase of amyloid deposition increasingly commands researchers’ attention. Does having “a head full of amyloid” mean a non-demented person is heading toward AD? What factors play into this question? This is where multimodal studies come in.
First, an attempt to account for cognitive reserve. Numerous studies have pinned a degree of protection from AD on education and intelligence. Dorene Rentz of Massachusetts General Hospital, Boston, shared the latest data on 84 non-demented research volunteers in their seventies who had a PIB-PET scan at baseline. At that point, they scored a perfect 0 or a 0.5, indicating very mild/prodromal symptoms on the Clinical Dementia Rating (CDR) scale and then underwent subsequent neuropsychological testing over some 18 months to date of follow-up. In Miami, Rentz showed results of multivariate modeling with those data, which sought to unpack the effects of amyloid and cognitive reserve.
Having amyloid in the precuneus/posterior cingulate areas of the brain at baseline predicted that the person would subsequently decline in AD-relevant cognitive domains. Overall, people with high cognitive reserve withstood brain amyloid better than people with low cognitive reserve. However, within those groups, Rentz spotted some differences. People positive for brain amyloid who had high cognitive reserve declined mostly in memory tasks, particularly immediate and delayed recall. People positive for brain amyloid with low cognitive reserve tended to fall off on a broader range of tests. This included memory for cued and delayed recall such as is used in the Dubois criteria for prodromal AD (e.g., the free and cued selective reminding test, FCSRT), and visuospatial perception. Regardless of their cognitive reserve, all volunteers who had low brain amyloid improved with repeated testing, whereas volunteers with high amyloid showed no such practice effect. At the same time, regardless of amyloid, highly educated people were more cognitively stable than less educated fellow volunteers.
In short, the take-home message at this early point of this prospective study is that mounting levels of amyloid do portend cognitive decline, but high education shields against that. This means that competing factors at work in a given person complicate the relationship between amyloid and cognitive decline. And reporting from a longstanding longitudinal study of highly educated, otherwise healthy people that is now incorporating florbetapir PET scans, Karen Rodrigue from the University of Texas at Dallas correlated increasing cortical amyloid with a slowdown in processing speed.
Following amyloid deposition in longitudinal cohorts is necessary for many reasons. One is that cognitive reserve is one likely reason for the great variability researchers see in how well people tolerate brain amyloid. It could explain the nagging puzzle of a group of people in aging studies that Susan Resnick of the National Institute on Aging in Bethesda, Maryland, brought up: namely those who die without a known history of cognitive decline but upon autopsy prove to have had AD per the disease’s pathological definition. This finding lingers as a criticism of the amyloid hypothesis. Since it is a postmortem finding, it’s unclear if the person’s amyloid developed many years or shortly before death. Had the person lived, how long until dementia set in? A year? A decade? In discussion, other scientists cautioned that such studies sometimes apply a low pathological threshold to declare AD and that the cognitive instruments in some older longitudinal studies fail to pick up the subtle episodic memory problems of preclinical AD. More current studies looking at amyloid and other outcomes simultaneously do indicate that this pathology has consequences for cognition and even activities of daily living. For example, a poster presented at HAI by Gad Marshall of Massachusetts General Hospital found that among a group of 55 subjects with MCI or normal cognition from ADNI and an ancillary study, those who had the most brain amyloid also were the most impaired in high-level skills used in everyday tasks (such as shopping, handling finances), even after controlling for the impact of memory impairment. These results were driven by the MCI subjects.
In between amyloid deposition and the ultimate outcomes of cognition and daily function lies the big territory of what might be the neural substrates for any damage downstream of amyloid. This question has an existing literature of toxicity pathways from cell-based and animal model studies, but bringing human imaging to bear on it is largely unexplored space. Early forays into the new world of combined amyloid, FDG-PET, and fMRI connectivity imaging were on display in Miami. See Part 3 of this series.—Gabrielle Strobel.
This is Part 2 of a seven-part series. See also Part 1, Part 3, Part 4, Part 5, Part 6, Part 7. View a PDF of the entire series.
No Available Comments
At the 5th annual Human Amyloid Imaging conference held in Miami, Florida, on 14-15 January 2011, one of the hottest topics was what’s beginning to emerge from multimodal imaging studies that are aiming for a more comprehensive view of what happens in the brain once amyloid deposits there. To be sure, it’s early days for that; hence, some results cut opposite ways, data can be provocative but numbers small, and methods not fully established, much less standardized. But it’s where the bleeding edge of amyloid research is at right now, so here for your thought and inspiration are brand-new slices of data and discussion.
Stefan Foerster of the Technical University in Munich, Germany, made a preliminary case that amyloid jams neuronal networks. He noted previous work showing that the anatomical pattern of amyloid deposition as measured by PIB-PET, and hypometabolism as measured by FDG-PET, partly overlap both with each other and with the default-mode network of functional connectivity. In a new collaborative study, 20 patients with very mild AD and 15 controls, all in their sixties, underwent a PIB and a FDG-PET scan at baseline and again 27 months later. The scientists analyzed these images using a method called statistical parametric mapping, by which they can compare digitized images voxel by voxel in a fashion unbiased by previous assumptions. What did the scientists find? The baseline amyloid pattern and the follow-up hypometabolism pattern were the most similar. At baseline the amyloid was broad and did not expand in time; the hypometabolism pattern was smaller at baseline but spread over the next 27 months to then overlap very well with baseline amyloid deposition, Foerster said. In essence, this would imply that hypometabolism of neurons follows amyloid deposition in their area with some delay in time.
To get a sense of which networks the most amyloid-prone brain areas normally engage, the researchers took the regions with peak levels of PIB retention in their AD patients at baseline as "seeds" and calculated their functional connectivity networks in a separate dataset of 27 volunteers in their twenties who had undergone resting-state fMRI scans. Intriguingly, Foerster said, the networks normally subserving those brain areas most vulnerable to amyloid deposition overlapped even better with the actual areas of FDG-PET hypometabolism in the AD patients at follow-up than did the default-mode network. Overall, this points in the direction that amyloid deposits in a given brain region disrupt neurons locally and also disrupt its functional connectivity network, and some years later this shows up as hypometabolism, i.e., neuronal dysfunction in those areas. “These findings support the amyloid hypothesis,” Foerster said.
On a poster, Liang Wang of Massachusetts General Hospital, Boston, showed roughly similar trends in a study of 42 cognitively normal older volunteers who had resting-state connectivity MR scans and PIB-PET scans. Wang used different methods in a different study design. But similar to Foerster, he also saw that when a given region’s amyloid burden was high, intrinsic activity tended to be incoherent and break down both locally and long range across nodes of the default network.
Foerster’s talk prompted both compliments and caveats. For example, Cliff Jack of the Mayo Clinic in Rochester, Minnesota, whose colleagues are working on this as well, cautioned that the temporal ordering of "amyloid-then-dysfunction" does not hold true across the brain. “Some brain areas have amyloid deposition early but do not become dysfunctional until late in the disease. Only some areas are connected in this way, where early amyloid deposition correlates soon after with hypometabolic networks,” Jack said. Foerster readily acknowledged that he was putting out a provocative concept to stimulate discussion and further study.
Consider another study on what might lie between amyloid deposition and cognition. Patrizia Vannini of Brigham and Women’s Hospital in Boston won the 2011 HAI Young Investigator Award for her talk about what amyloid does to one particular brain area indispensable for episodic memory that lies smack in the default-mode network. The posteromedial cortex drew Vannini’s attention because it is not only prone to early amyloid deposition, but also figures in both encoding and retrieving memories. In what’s called the "encoding/retrieval flip," this area suppresses its default-mode firing during memory encoding and activates during cued retrieval. Perhaps, the BWH researchers reasoned, this area is so prone to amyloid deposition exactly because it is constantly active in these ways, that is, produces a lot of Aβ? This would jibe with a discovery by Randy Buckner of MGH that something about the activity and the metabolism of connectivity hubs is conducive to amyloid deposition (Buckner et al., 2009; see also Hedden et al., 2009), and fit with the finding that neural activity drives up Aβ secretion (Cirrito et al., 2005).
Vannini tested this hunch with an associative memory task for which 26 young and 41 old volunteers—half of them with, half without brain amyloid—lay in the scanner and tried to memorize which name went with which face flashed on a screen. Later, they were tested on how well they did. Initially, Vannini found that old people were less able than young people to "flip this switch" in brain activity. A bit like gymnasts who lose flexibility, their posteromedial flips between deactivation and activation happened within a smaller dynamic range. When she split the old volunteers by whether they had brain amyloid, she found that brain amyloid further limited this ability to modulate. Furthermore, people who modulated, i.e., "flipped," well tended to get the test right. This would mean that being able to mount a nimble, dynamic response in the posteromedial cortex is important for memory, and both age and amyloid get in the way of that.
Other scientists, too, are probing how brain activation during memory encoding changes as people age, and what, if anything, amyloid has to do with that. Elizabeth Mormino of the University of California, Berkeley, first pointed to discrepancies between amyloid and cognitive status from person to person. How can it be that some older people with high amyloid function better than others? Mormino investigated a hunch that the former manage to call on the prefrontal cortex to compensate for the presence of amyloid. She studied 37 people in their seventies and 15 in their twenties in a more complex recognition test in which volunteers see landscape pictures while lying in the scanner and later, outside the scanner, get tested on whether they had actually encoded the picture. In this way, Mormino and her colleagues visualized the cortical areas that were active while volunteers encoded what they saw. Roughly similar to Vannini, Mormino also saw greater activation in young than in old subjects, and a trend for further reduction with amyloid.
One result stood out. People with brain amyloid activated their prefrontal cortex significantly more than those without. “This was a striking pattern,” Mormino said, and added, “It is possible that the integrity of the prefrontal cortex enables these subjects to remain in the normal range despite having amyloid.” Perhaps people whose prefrontal cortex escapes other insults that can accompany aging—vascular disease, for example—withstand the degrading effects of amyloid longer than do their fellow elders with a less preserved prefrontal cortex. This, then, could be one potential explanation for how some people "live in peace with their amyloid" for some years.
It sounded appealing until the next two speakers presented what appeared to be the opposite result. Kristen Kennedy of the University of Texas at Dallas reported on an fMRI/amyloid PET study of 137 adults aged 30 to 89 who had an AV-45 (aka florbetapir/Amyvid) scan and performed essentially the same memory encoding task Mormino’s group uses. Here, older people with high amyloid activated their prefrontal cortex less during encoding than did amyloid-free age-matched fellow volunteers. In Kennedy’s hands, people with brain amyloid performed worse, and show a twin pattern of reduced prefrontal activation and failure to suppress the default-mode network as has been shown previously (Hedden et al., 2009).
Michael Devous, also of the UT group in Dallas, presented functional connectivity MR data available to date from the Dallas Lifespan Brain Study on the default-mode network, which is active at rest, and the salience network, one of many networks active during focused tasks. Overall, his work indicates that when amyloid deposits in the brains of cognitively normal people, connectivity in the default-mode network falls apart, whereas in the salience network, it shifts, such that some areas increase abnormally. In particular, Devous noted seeing a steady destruction of the prefrontal cortex components of the default-mode network in people with high brain amyloid. MGH’s Wang, too, had noted a disruption of connectivity in the prefrontal cortex in cognitively normal people with more amyloid.
Finally, a decrease in prefrontal activity in normal people with brain amyloid also showed up in the work of Prashanthi Vemuri of the Mayo Clinic in Rochester, Minnesota. Vemuri studies how functional connectivity changes along the spectrum from cognitively normal to overt AD. Vemuri’s group did PIB-PET imaging and resting-state fMRI in some 250 folks who participate in the longitudinal Mayo Clinic Study of Aging. She grouped them as cognitively normal with or without amyloid, MCI with amyloid, and AD. She excluded people with MCI but without amyloid, “because we wanted a pure group on the AD path.”
Viewed globally, Vemuri saw functional connectivity in cognitively normal people with PIB go up, possibly reflecting that the brain reorganizes its functional networks to compensate for the presence of amyloid pathology. In people who were progressively further along the path to AD, functional connectivity was lower. Vemuri analyzed the brain’s "connectome," i.e., multiple functional networks, to look for changes within and among 25 major networks that she had previously identified in volunteers without amyloid. In this way, she saw functional rearrangements at the early stage (i.e., disconnection of the frontal and temporal-parietal subsystems) and consistent decreases within and across networks at the later stages.
These data are complex to interpret, though in Vemuri’s mind it still boils down to some simple messages. For one, the results in cognitively normal PIB-positive people reflect what amyloid alone does to connectivity, whereas the results on the MCI/PIB-positives and AD groups reflect what both amyloid and neurofibrillary pathology do together. For another, early on, while only amyloid pathology is at play, the two major subsystems affected—the frontal and the temporal/parietal cortex—react differently, with decreased connections with each other and increased connections within each subsystem. Once both pathologies are there, connectivity decreases overall consistently.
What gives? In discussion, scientists challenged each other as to what this data amount to when one area of focus, the prefrontal cortex, goes up in one study and down in another using the same fMRI paradigm. The studies were all different in how they judged a volunteer to be amyloid positive, which amyloid tracer was used, what sorts of people were enrolled, how ApoE might affect the outcomes, how the fMRI data were analyzed, and where researchers placed the seed region to visualize various networks. Moreover, noise, non-specific binding, and atrophy effects can blur data on small effects, cautioned Bill Jagust of UC Berkeley. “Connectivity networks are complicated to study. Given how nascent this field is, our interpretation must be cautious at this point,” Jagust told ARF. For this reason, scientists agreed, the field would do well to continue for some time longer with exploratory studies to let the technical issues play out, and not try to standardize methods just yet. (In contrast, standardization is beginning in MRI and well underway in CSF methodology for AD research.)
There was some disagreement about why the cognitive effects in these studies are so subtle. Some researchers found it “shocking” that the amyloid effect on cognitive measures in these studies is small. Others replied that they are small by definition because the studies enroll cognitively normal people. “Don’t forget that most patients with large effects have AD and amyloid,” said Reisa Sperling of Brigham and Women’s Hospital in Boston. The mystery, all agreed, lies in why some people can stay cognitively normal with amyloid in their brains for years.
Scientists also discussed how they could move from correlation to cause. “Right now, we have associations among amyloid, age, education, cognition, activity. How do we design studies so we can turn these associations into mechanistic relationships?” asked Steve Arnold of the University of Pennsylvania in Philadelphia. The answer, others said, lies in multimodal longitudinal studies such as ADNI, AIBL, DIAN, and increasingly others as longitudinal aging studies are bulking up on neuroimaging.
While multimodal imaging in AD research is young, the larger field of aging research has been transformed by the advent of amyloid PET. “Human amyloid imaging helps us parse which parts of aging are due to amyloid and which are due to other age-related insults,” said Sperling. One example for this was a functional imaging poster by Trey Hedden and colleagues at MGH. It showed that a significant fraction of older cognitively normal people was unable to dynamically modulate brain activity such that they would devote more attention to increasingly difficult tasks, and it ascribed this executive deficit to white matter hyperintensities, not to amyloid.
The overall change to the field has been “momentous,” said Randy Buckner of MGH in a keynote address, particularly when amyloid imaging is viewed together with other biomarkers. Collectively, these markers now enable scientists to view the progression of the disease in living people, Buckner said, and they have made explicit the staging model for the disease that was forming in people’s minds (see Jack et al., 2010; Perrin et al., 2009). “From this emerges our current focus on using these markers in asymptomatic individuals,” Buckner said. As this research advances, the staging model may shift, Buckner noted. Amyloid will stay earlier than atrophy, but the cognition curve may move to the left as scientists get better at detecting small impairments.
For a panoramic view, Bucker said he is inspired by a conceptual framework whereby metabolic conditions in early life set the stage for amyloid deposition in midlife, which itself precedes atrophy and then dementia in late life. These metabolic conditions are not yet understood, but aspects of glucose metabolism and mitochondrial function likely play a role (e.g., Valla et al., 2010; Vlassenko et al., 2010). By this framework, genetic risk factors such as ApoE and perhaps Tomm40 affect not so much the neurodegenerative phase late in life, which is the context in which they are frequently studied in experimental bench research. Rather, genetic risk factors primarily affect how a person’s brain gets wired up, Buckner suggested. In short, genetics define vulnerability by the time of young adulthood; the rest—metabolism, hubs, activity patterns, amyloid, tau, atrophy, cognition—unfolds from there.—Gabrielle Strobel.
This is Part 3 of a seven-part series. See also Part 1, Part 2, Part 4, Part 5, Part 6, and Part 7. View a PDF of the entire series.
No Available Comments
Held 14-15 January 2011 in Miami, Florida, the 5th annual Human Amyloid Imaging conference illustrated how this form of PET imaging is becoming broadly embedded throughout human Alzheimer’s disease research. Consider these examples.
Agneta Nordberg of the Karolinska Institute in Stockholm, Sweden, showed that amyloid imaging, in this case with 11C PIB, forms part a multi-tracer study by which this group is following patients with sporadic AD, MCI, and also families with autosomal-dominant mutations in APP and presenilin. Reminiscent of DIAN, this study measures neuropsychology, protein biochemistry in the CSF, brain structure with MRI, and brain metabolism with FDG-PET. The new piece Nordberg introduced in Miami was an attempt to see how neuroinflammation plays into AD by adding on PET imaging with 11C-d-deprenyl. This tracer visualizes astrocytosis, said Nordberg.
Inflammation is of intense interest in AD research, but has remained largely elusive to brain imaging. Some studies have used the only available PET tracer for neuroinflammation—(R)-PK11195 for microglia—together with PIB (Wiley et al. 2009; Okello et al., 2009; Yokokura et al., 2011; see also Schuitemaker et al., 2010), but many AD researchers consider PK11195 unsatisfactory. As for 11C-d-deprenyl, little is known about it. Nordberg mentioned recent work on this tracer in amyotrophic lateral sclerosis, Creutzfeldt-Jakob disease, and epilepsy, all indicating that astrocytosis happens early in disease. Arthritic joints have been reported to take it up (Danfors et al., 1997).
Comparing PIB and d-deprenyl retention in the same volunteers, Nordberg’s studies are ongoing. To date, her team sees that d-deprenyl tends to go up early, though amyloid and this astrocytosis marker appear not to be regionally correlated. D-deprenyl retention was high in MCI patients who are PIB-positive; it was also high in mutation carriers who are cognitively normal and whose amyloid burden is low. It was relatively lower in people who had advanced to Alzheimer’s, suggesting that astrocytosis appears to happen early on at the MCI or even pre-symptomatic stage. “This study shows we can do multimodal imaging studies to look at more variables at once in the same patients. This marker could potentially be incorporated into drug testing,” Nordberg said. “We are very excited about this and are trying to recruit more patients participating in this study,” she told ARF. That said, d-deprenyl does not image neuroinflammation exhaustively; more markers are needed.
On that note, Nordberg mentioned that amyloid imaging of patients with the Arctic mutation reinforces a related point that is sometimes overlooked as the first amyloid tracers are close to receiving regulatory approval for clinical use (see ARF related news story). That is, scientists urgently need PET tracers to image oligomeric/protofibrillar forms of pathogenic proteins to fill in the picture of what is going on in the brain. Case in point: Nordberg showed PET images of a 60-year-old woman who has AD resulting from the Arctic APP mutation, and this woman is PIB negative. The Arctic mutation causes early onset AD that progresses rapidly but is otherwise clinically quite typical. Rather than the classic cored, fibrillar amyloid plaques, this form of AD turned out on postmortem pathology to have caused ring-shaped plaques without a distinct core (Basun et al., 2008). The mutation does cause abundant formation of smaller protofibrillar forms of Aβ. These cases are extremely rare; however, they show that clinical AD can arise from forms of Aβ that neither of the current fibrillar amyloid tracers pick up. Imaging these smaller forms of Aβ would add much to understanding the disease. “We need more PET ligands,” Nordberg said.
A poster on antidepressants and brain amyloid created a buzz at HAI as well. Yvette Sheline of Washington University, St. Louis, Missouri, showed that among 186 cognitively normal elderly research volunteers who had undergone a PIB-PET scan, those who had taken antidepressant medication in the past five years had less brain amyloid. The longer these volunteers had been taking antidepressants, the less amyloid they had. The volunteers were otherwise closely matched in age, ApoE and other factors that might affect their amyloid load. The antidepressants in question here are the selective serotonin reuptake inhibitors. The human studies were prompted by earlier results in mice. Using microdialysis, Sheline’s coauthor John Cirrito and colleagues had found that both citalopram and fluoxetine, as well as direct infusion of serotonin, slashed soluble brain Aβ levels by a quarter to a third. This suggests that serotonin signaling might affect amyloid deposition, Sheline said.
How this works remains unclear, as this line of research is at an early stage. The mouse studies were acute, not long-term. The human data came from a so-called sample of convenience, people who were asked about their antidepressant use, not from a drug trial designed to test this hypothesis. Only one such trial has published a reduction of PIB retention to date (Rinne et al., 2010; see also Part 5 of this series). Yet given the well-known safety profile of SSRIs, researchers might want to consider randomized trials to see if these drugs indeed could double as amyloid-lowering agents. At this point, Sheline’s message to readers is: Don’t try this at home.
Consider one more example of amyloid imaging in AD research. Keith Johnson of Massachusetts General Hospital, Boston, used it to take a closer look at microbleeds. These small ruptures in a blood vessel have caused concern in AD clinical trials and have generated a literature in mouse models, but remain poorly understood. The tiny dollop of blood eventually turns into hemosiderin, which makes a big black dot on MRI. A population-based imaging study had pinned their prevalence at about 30 percent by age 75, and had suggested that microbleeds seen in the brain’s upper, i.e., lobar regions, are related to CAA and AD, whereas microbleeds occurring deep inside the brain have to do with cerebrovascular disease (Vernooij et al., 2008).
Johnson and colleagues looked at both microbleeds and CAA together with an MRI/PIB/FDG-PET study in 92 cognitively normal or very mildly impaired people in their seventies, who also underwent cognitive tests and genotyping. When two raters analyzed the images separately, it became clear that distinguishing them from vessels that appear cut across in the plane of view was not trivial. “You have to be careful not to overcall them,” Johnson said.
How common were these signs of vascular injury in the brains of this set of older people? Twenty-three of the 92 folks had microbleeds, about evenly split between lobar and deep. Of the 23, most people had one microbleed, a few had two, two people had four or five. What were they related to? To ApoE status and age, to cortical PIB retention in the case of lobar microbleeds, and to a diagnosis of hypertension in the case of deep microbleeds. In other words, older people with ApoE4 who had amyloid deposition or high blood pressure were most likely to have a microbleed. Was having a microbleed in any way related to cognitive performance or cerebral hypometabolism? Not in this sample, Johnson reported. “In normal people, the microbleeds are somewhat common. They are a feature of preclinical AD but appear not to be associated with significant clinical findings,” Johnson said.
On balance, these data sounded less alarming than some previous studies; however, Johnson pointed out that the MGH sample was smaller and healthier than the Rotterdam cohort, for example. The HAI audience followed this talk with an animated discussion. The underlying idea is that vascular amyloid increases the risk for bleeding. Hence, when a microbleed shows up on MRI, that implies the person might have CAA and be at higher risk for stroke. But how that is interpreted in clinical practice varies quite a bit. Some centers go so far as to infer a person probably has AD when a microbleed shows up on MRI. Other scientists said this was jumping to conclusions and should not be done. Other practitioners assume that when they see one microbleed, it represents the tip of the iceberg. Here, too, Johnson’s data would suggest that is wrong. Johnson noted that his group used a highly sensitive technique and scrutinized the brains quite exhaustively. “My hunch is that there aren’t automatically many more if you see one,” Johnson said. Charles DeCarli of the University of California, Davis, an expert on vascular dementia, concurred. “People sometimes overestimate the import of a microbleed,” DeCarli said. “In a community setting, about 70 percent of people in this age group have single silent infarcts. On the cognitive side, these silent infarcts do not do much. When you have a PIB-positive person with a microbleed, that is probably CAA. But you cannot simply reverse this implication.”—Gabrielle Strobel.
This is Part 4 of a seven-part series. See also Part 1, Part 2, Part 3, Part 5, Part 6, and Part 7. View a PDF of the entire series.
No Available Comments
Kenji Ishii of the Tokyo Metropolitan Institute of Gerontology updated the audience at the 5th annual Human Amyloid Imaging conference, held in Miami, Florida, on 14-15 January 2011, about where the Japanese ADNI stands at this point. His talk reinforced how age and genetics influence amyloid formation and AD. Responding to unexpectedly high interest from its participants, the Japanese ADNI ended up scanning 450 people with amyloid PET, Ishii said, three times as many as they had originally planned. The resulting data are compatible with those from the U.S. ADNI and the Australian AIBL study without much of an ethnic confound, Ishii said. This means J-ADNI adds a cohort for combined analyses of larger datasets.
There are differences, though. The normal controls in the J-ADNI cohort are about a decade younger than its U.S. counterpart, and have fewer ApoE4 carriers among them. Consequently, only 21 percent were amyloid positive, less than half the rate of the U.S. ADNI controls. Similar to U.S. cohorts, though, in the Japanese volunteers, tracer retention started rising in the late sixties in people without an E4 allele, earlier with one, and earlier still with two. The PIB positivity rate tracked that: Around age 60, it was only 5 percent in those without ApoE4, but 37 percent in those with one copy and 67 percent for people with two copies. “ApoE accounts for at least a 10-year age difference in amyloid deposition,” Ishii said.
A note on 18F amyloid tracers and histopathology correlations. While Avid and Lilly learned on 20 January at the Food and Drug Administration that their application for clinical use of Amyvid—the most advanced 18F compound in the running—will be approvable pending further data on reader training and consistency (see ARF related news story), the Miami conference featured new data on what the runners-up are doing and how well PET tracers in general detect fibrillar amyloid pathology in the brain.
Steve Arnold of the University of Pennsylvania, Philadelphia, reported that 18F-flutemetamol, the fluorine-18 version of PIB being developed by GE Healthcare, correlates well with histopathological amyloid as measured in biopsy samples. Brain biopsies are rare, but people with normal pressure hydrocephalus sometimes have a small piece of tissue taken out when surgeons place a shunt from a brain ventricle to the peritoneum to let excess fluid drain. (This therapeutic procedure came up at last Thursday’s FDA Advisory Committee meeting on Amyvid, as well, when scientists entreated the committee to recommend approval by citing a range of examples for how amyloid imaging would improve their clinical care. One such instance is hydrocephalus. Patients with this condition tend to respond well to shunting if they do not also have amyloid pathology, but poorly if they do, so amyloid PET would enable clinicians to pick the patients for whom this invasive treatment is right.)
In Miami, Arnold presented data on seven hydrocephalus patients who’d had a shunt placed, a biopsy taken, and a flutemetamol scan thereafter. Staining of the fixed specimen with antibodies to various fibrillogenic proteins and with thioflavin S showed that four of the seven people indeed had Aβ amyloid in their brains, and one had CAA as well. This matched entirely with the flutemetamol scans, Arnold said. Seven cases don’t make much of a quantification, but still, this is the first study to look for concordance of flutemetamol imaging with histopathological evidence of Aβ-related lesions, Arnold said, and it fits with the existing literature on PIB matching up against pathology. Juha Rinne of the PET Center in Turku, Finland, presented a similar, but larger biopsy study for hydrocephalus cases using PIB scans. This work reinforces again that this tracer correlates strongly with the number of Aβ aggregates in the respective brain areas. And on pathology-PET tracer correlation in general, outside of biopsies, Ira Driscoll, at the National Institute on Aging in Baltimore, Maryland, presented her results of six additional autopsy cases. In her hands, the tracer she tested, PIB, accurately reflected the amount of Aβ pathology in different brain regions. “PiB does a good job of capturing Aβ in vivo,” Driscoll later wrote to ARF.
On a poster, Milos Ikonomovic of the University of Pittsburgh Medical School showed biochemical and pathology data to suggest that flutemetamol retention in the brain reflects Aβ plaque load similar to PIB. This tracer is beginning to be more widely used in research settings as well. Ranjan Duara of Mount Sinai Medical Center in Miami Beach, Florida, showed data on using flutemetamol together with MRI for classifying different kinds of MCI patient. G.E. is currently performing a Phase 3 multicenter histopathology study.
Also on flutemetamol, Rik Vandenberghe of K.U. Leuven, Belgium, and colleagues presented research on automated software and on a separate supervised machine learning technique applied to the scans from a Phase 2 study (Vandenberghe et al., 2010). The ultimate goal is to use machines to interpret scans instead of relying on human raters. It was around consistency among raters in deciding whether a given amyloid scan is positive or not, especially in the early disease stage, that Amyvid’s bid on 20 January for FDA Advisory Committee approval ran into a temporary snag, even though the company had followed a statistical plan the FDA had previously okayed. Getting this call right in the clinically relevant population is a pressing concern now. In Miami, several speakers, including Wenzhu Bi of UPitt and Adam Fleisher of the Banner Alzheimer’s Institute in Phoenix, Arizona, presented methods to that end, namely to set thresholds for positivity of a scan with PIB and florbetapir, respectively.—Gabrielle Strobel.
This is Part 5 of a seven-part series. See also Part 1, Part 2, Part 3, Part 4, Part 6, and Part 7. View a PDF of the entire series.
No Available Comments
The 5th HAI conference, held in Miami Beach, Florida, concluded on 15 January with a panel discussion of how far amyloid PET has come in the seven years since its first major publication (Klunk et al., 2004) when viewed from the perspective of therapy development. After two days of talks on an expanding breadth of basic science studies with these tracers, Eric Siemers of Eli Lilly and Company summed up the theme by issuing this call to his colleagues: “Don’t get too caught up in the 'gee whiz' of amyloid imaging—and it is fascinating science!—when the goal is to use this to improve clinical trials and make a difference in the treatment of the disease.” To that end, what do scientists know? Which trial purposes are amyloid imaging well suited to accomplish, what can’t it do? Siemers; Howard Feldman of Bristol-Myers Squibb; Michael Grundman, formerly of Elan and now at Global R&D Partners, San Diego; and Paul Aisen of the Alzheimer's Study Group, also there, weighed these questions before the crowd dispersed for the airport, the beaches, or Miami’s hip Art Deco district, as the case may be. Here are the main points.
In the past few years, scientists and clinicians in AD have coalesced around a consensus that amyloid deposition precedes the traditional diagnosis of AD by around 15 years. As cognitive measures become more sensitive and diagnostic criteria evolve, this number shrinks somewhat, but it still leaves a long time for neurodegeneration to eat away at the brain and make a case for intervening early.
Feldman first said that clinicians had put much effort into reframing the concept for the years before frank dementia, and then he placed amyloid imaging into this concept. In a nutshell, trialists are pushing leftward on the disease’s timeline. They have defined the stage immediately prior as prodromal AD (this would capture the large fraction of people with MCI who progress to AD) and the stage before that as asymptomatic at-risk (those are cognitively normal people with biomarker changes; see Dubois et al., 2010; Dubois et al., 2007). A separate set of new diagnostic guidelines is substantially similar, most scientists agree, with differences largely in semantics (see ARF ICAD news story).
Trials in the prodromal and the asymptomatic at-risk groups are new ground, Feldman said. For them to happen, biomarkers need to accomplish several things. Biomarkers need to identify people with brain amyloid. AIBL, ADNI, and other studies showed that about a third or more MCI patients have no or low brain amyloid. These people should not be in the same trial as, or at least not be analyzed together with, people with abundant amyloid. It seems clear that both amyloid PET and CSF assays can do that. Avid’s Phase 3 histopathology study shows that florbetapir detects aggregated forms of brain amyloid, and the ADNI CSF team showed that it is possible to set a cutoff point for Aβ and tau that distinguishes healthy controls from AD in postmortem series, Feldman said.
Further, biomarkers need to ascertain that trial participants will move along the continuum to AD dementia. For progression from the prodromal stage to AD dementia, converging evidence from multiple studies portends that CSF predicts progression, and a florbetapir Phase 2 trial also shows faster progression in amyloid-positive participants. Moreover, CSF biomarkers and amyloid PET correspond well to each other (Fagan et al., 2006; Jagust, 2009). “Again, both amyloid markers can do this,” Feldman said. Data on the progression from the asymptomatic at-risk to the prodromal stage is a bit further behind but heading in the same direction.
With this relentless focus on early-stage trials, what about the current patients? Will they be forgotten after one trial after another in them has failed for the past decade? Not at all, the panel agreed, and biomarkers will hopefully help give trials in this phase a better shot at success, as well. It is generally accepted that about every fifth person diagnosed as having AD turns out after death not to have had amyloid pathology. If AD is defined by its signature pathology, Feldman said, then these 20 percent of cases should not be called Alzheimer’s, and should not enroll in those trials. For his part, Siemers added that in the course of semagacestat testing, some 15 percent of 600 trial participants who had florbetapir scans turned out not to have amyloid. “That will dilute your trial,” Siemers said.
The panel agreed easily, then, that the time has come for using amyloid markers to select patients for trials in all stages of AD. The question of whether these markers make good outcome measures is more difficult. In theory, amyloid imaging could serve as an endpoint, said Grundman. It could do any of this: show that the drug reaches and affects the target, tell how quickly the drug does this, where in the brain this happens, help with dose selection, help decide whether to continue or abandon development of a given treatment, and tie the target engagement to a subsequent clinical outcome, i.e., the result that ultimately matters to the patient. How did amyloid imaging fare on these challenges when it was put to the test in a small PIB-PET Phase 2 Bapineuzumab trial (Rinne et al., 2010)? The treatment did engage the target (amyloid went down a bit in people on antibody, up a bit in people on placebo), this happened slowly (over 78 weeks), and amyloid went down in all regions evaluated. On the remaining points, this one study alone provided no answers.
Despite these early hints, scientists don’t have their ducks in a row on outcomes nearly as well as on enrichment. For example, in Rinne et al., PIB uptake went down, but CSF Aβ42 did not. Similarly, in AN1792’s Phase 2, amyloid pathology went down as per postmortem pathology, but CSF Aβ42 did not. In AN1792, CSF total tau went down; in the Bapineuzumab Phase 2, it did not, though phospho-tau showed a trend. With semagacestat, there was no effect on CSF Aβ or tau in the trial, but a separate study done with the Silk method of continuous CSF monitoring did show that effect for therapeutic doses used in the trial (Bateman et al., 2009). In a Phase 2 trial, PBT-2 lowered CSF Aβ42 but not tau.
Clearly, the field has not validated an amyloid endpoint as an outcome measure. “If you take a landscape view, it is that we struggle to understand the nuances of what these various results are telling us. We will have to reconcile these differences between CSF and PET. We are certainly not out of the woods,” Feldman said. On the million dollar question of how amyloid correlates with clinical benefit, Aisen urged the field to keep an open mind. “If, indeed, Aβ oligomers are the most toxic species, then in theory, a drug that increases PIB might be beneficial. The answer is likely a long way off,” Aisen said.
All agreed that it may take many trials to sort these questions out. Where to start? Aisen, who, as head of The Alzheimer's Disease Cooperative Study (ADCS), can speak more freely than his industry colleagues about trial designs he has up his sleeve, suggested a design that presented itself out of a recent analysis of ADNI data. It turns out that the normal controls fell into an amyloid-negative and an amyloid-positive group. What’s more, as observation went on, these groups clearly diverged on brain atrophy and ventricular enlargement, and showed a difference on cognition even on a measure as crude as the MMSE. Together, those differences among the "normals" are big enough to support a trial, and power estimates based on them call for group sizes of 300. “That is entirely doable,” Aisen said. Consequently, the ADCS is proposing a trial to the National Institute on Aging. It aims to screen cognitively normal people in their seventies, either with a lumbar puncture or amyloid PET, and would treat the amyloid-positives among them with an anti-amyloid treatment for two years. The trial would measure a fleet of outcome measures including MRI, FDG-PET, sensitive cognitive tests using free or cued selective reminding, and amyloid PET.
This would be secondary prevention. The planning happening at this point in time would constitute groundwork for more such trials. The price tag for amyloid PET will influence how much of this groundwork can be realized, scientists agreed. Screening with this technology may seem prohibitive to a society strapped by deficits and budget cuts in towns, states, and at the federal level; however, Feldman pointed out that primary prevention trials are much larger and more expensive. “In comparison, this seems manageable,” he said.
In the day’s discussions, these points came up:
Question: How will you select what you test in these secondary prevention trials? Nothing has worked yet!
Aisen: There are two basic ideas about that: the safety and the efficacy rationale. Some suggest we should pick an extremely safe drug because it is an asymptomatic population. I am in the other camp, which emphasizes the scientific rationale behind the drug and clear data that the drug hits its target. If we launch this with NIA funding, we will ask an external panel to help us select. It will be an imperfect process.
Reisa Sperling, BWH, Boston: In selecting a drug, we do need evidence of target engagement, but we should not require clinical signals, i.e., an effect in mild to moderate AD. In cancer and heart disease, some drugs help early but not late.
Question: will the FDA accept this?
Aisen: The FDA and EMA support trials with biomarkers anchored by some subtle cognitive marker for familial AD. The bar may be higher for the population I envision, but I see regulatory acceptance for this even for LOAD on the horizon.
Question: What about cognitive reserve?
Adam Fleisher, Banner Alzheimer Institute, Phoenix, Arizona: The consequence of amyloid varies strongly by cognitive reserve. We do not understand the relationship between the amount of amyloid and whether it predisposes to clinical symptoms very well, so we can’t define the boundary between normal aging and AD in this way. But we know in general that if you have elevated amyloid, you are at higher risk of developing AD.
Sperling: Within the group of amyloid-positive normals, there might be a mix of people who are on their way to AD and other people who are good at withstanding the effects of amyloid. If that is so, then we need a way to split these to make sure we are not enrolling the people with greatest reserve into the trials.
The HAI conference then ended with a dual message: Stephen Salloway from Butler Hospital in Providence, Rhode Island, coupled kudos to Bill Klunk and Chet Mathis for opening up this new field with a plea to all investigators to also keep track of the patients who don’t fit the concept. Those would be people who have amyloid but primarily suffer from a different disease, such as dementia with Lewy bodies, or people who have amyloid but also vascular disease, and people who have dementia but no amyloid. It may also include the oldest old, in whom dementia is caused by amyloid and a number of comorbid conditions, challenging clinicians to determine the relative contribution of each of these factors to the patient’s cognitive decline.—Gabrielle Strobel.
This is Part 6 of a seven-part series. See also Part 1, Part 2, Part 3, Part 4, Part 5, and Part 7. View a PDF of the entire series.
Below we list 66 of the 67 abstracts presented at the Human Amyloid Imaging Conference held 14-15 January 2011 in Miami, Florida. (One was withheld due to publication embargo.) All meeting abstracts are published in the Alzforum Papers of the Week database. The accompanying news series covers many but not all of these studies; hence, this document offers the curious reader a wealth of new data to peruse. The Alzforum editors thank co-organizer Keith Johnson for obtaining permission to publish the abstracts; we also acknowledge all the authors for their generosity in sharing their latest data with the worldwide Alzheimer 's disease research community.—Gabrielle Strobel.
Cohen AD, Price JC, Mathis CA, Nebes RD, Saxton JA, Snitz BE, Aizenstein HJ, Weissfeld LA, DeKosky ST, Klunk WE. Longitudinal changes in fibrillar amyloid-beta deposition across the cognitive spectrum. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15; Abstract
Rentz DM, Betensky RA, Becker JA, England RL, Maye J, Gidicsin C, Sperling RA, Johnson KA. Amyloid deposition in non-demented elderly predicts longitudinal cognitive decline. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Koivunen J, Scheinin N, Virta JR, Aalto S, Vahlberg T, Nagren K, Helin S, Parkkola R, Viitanen M, Rinne JO. PET imaging of beta-amyloid deposition in patients with mild cognitive impairment: a two year follow-up study. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Forster S, Grimmer T, Miederer I, Henriksen G, Yousefi BH, Greve D, Graner P, Wester H-J, Forstl H, Drzezga A. Regional expansion of cerebral hypometabolism in AD follows the pattern of amyloid-deposition with temporal delay and is related to a healthy functional connectivity network. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Mormino EC, Brandel MG, Madison CM, Jagust WJ. Increased prefrontal activation in amyloid positive cognitively normal individuals during successful episodic memory encoding. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Kennedy KM, Rodrigue KM, Hebrank AC, Devous Sr. MD, Park DC. Precuneus beta-amyloid burden is associated with decreased bilateral frontal activation and default network suppression in healthy adults. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Devous Sr. MD, Kennedy KM, Rodrigue KM, Hebrank AC, Park DC. Precuneus beta-amyloid burden correlates with altered cortical network function in a lifespan sample of healthy adults. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Vannini P, Hedden T, Sullivan C, Ward A, Becker JA, Johnson KA, Sperling RA. Age and amyloid deposition are associated with functional alterations in posteromedial cortex during encoding and retrieval processes. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Vemuri P, Jones DT, Machulda M, Przybelski SA, Boeve BF, Knopman DS, Petersen RC, Jack Jr. CR. Functional reorganization of the brain along the Alzheimer's disease continuum. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Nordberg A, Scholl M, Carter S, Kadir A, Almkvist O, Ng Y-B, Wall A, Engler H, Graff C, Langstrom B. Biomarkers for following Alzheimer's disease progression. Amyloid 11C-PIB imaging in a multi-tracer paradigm with 11C-d-deprenyl and 18F-FDG studied in preclinial and clinical AD. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Ishii K, Sakata M, Oda K, Toyohara J, Ishiwata K, Senda M, Ito K, Kuwano R, Iwatsubo T. Influence of APOE genotype on amyloid deposition in Japanese population. Direct comparison of J-ADNI, USADNI and AIBL 11C-PiB PET data. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Scheinin NM, Aalto S, Kaprio J, Koskenvuo M, Raiha I, Rokka J, Hinkka-Yli-Salomaki S, Rinne JO. Early detection of Alzheimer's. 11C-PIB PET in monozygotic and dizygotic twins discordant for cognitive impairment. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Rabinovici GD, Rosen HJ, Alkalay A, Trojanowski JQ, Grinberg LT, Huang EJ, Seeley WW, Miller BL, Jagust WJ. PIB versus FDG PET in pathologically verified dementia. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Wolk DA, Grachev ID, Buckley C, Arnold SE. 18F-flutemetamol amyloid imaging has strong concordance with cortical biopsy histopathology. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Driscoll I, Troncoso JC, Sojkova J, Zhou Y, Rudow G, Kraut MA, Ferrucci L, Mathis CA, Klunk WE, Resnick SM. Correspondence between in vivo PiB-PET amyloid imaging and post-mortem, regional burden of As and Tau lesions. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Johnson KA, Gidicsin C, Larvie M, Maye J, Carmasin J, Becker JA, Rentz D, Sperling RA. Microbleeds and amyloid-s burden in non-demented elderly. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Matthews DC, Andrews RD, Ordonez CE, Schmidt ME. Methodologic considerations in the acquisition and analysis of amyloid imaging data for pharmaceutical clinical trials. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Nelissen N, Van Laere K, Thurfjell L, Buckley C, Farrar G, Brooks DJ, Vandenberghe R. Support vector machine analysis of flutemetamol scans. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Bi W, Weissfeld LA, Cohen AD, Klunk WE, Mathis CA, Price JC. Exploration of the PiB positivity boundary using statistical clustering. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Landau SM, Price JC, Jagust WJ, Mathis CA. Reliability of longitudinal PIB: How do data processing methods influence detection of change over time? Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Tolboom N, Ossenkoppele R, Yaqub M, Boellaard R, Scheltens P, Lammertsma AA, van Berckel BNM. Longitudinal amyloid imaging using [11C]PIB: Choosing the right method. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Koeppe RA. Amyloid PET imaging in large multi-center trials: Technical and practical issues. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Xu G, Christian BT, Wooten DW, Hillmer AT, Murali D, Barnhart T, Harding S, Cleary C, Kastman EK, Johnson SC. The influence of amyloid burden on cerebral glucose metabolism and cognition in cognitively normal middle age subjects who have high risk for Alzheimer's disease. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Vlassenko AG, Couture L, Goate AM, Raichle ME, Morris JC, Mintun MA. Regional brain metabolism in cognitively normal elder individuals with various levels of cerebral beta-amyloid deposition. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Lee DY, Han Choo IL, Kim JW, Seo EH, Lee DS, Kim YK, Kim SG, Park SY, Woo JI. Relationship of amyloid-beta burden with age-at-onset in Alzheimer's disease. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Fleisher AS, Chen K, Roontiva A, Thiyyagura P, Liu X, Clark CM, Mintun MA, Pontecorvo MJ, Doraiswamy PM, Reiman EM. Multi-level fibrillar amyloid thresholds of florbetapir F18 PET images from five multi-center studies. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Langbaum JB, Chen K, Fleisher AS, Keppler J, Prouty A, Bandy D, Perkins M, Richter NK, Jakimovich L, Reiman EM. [18F]AZD4694 in the symptomatic and presymptomatic study of Alzheimer's disease Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
van Dyck CH, Bruck A, Barcelos NM, Planeta-Wilson B, Benincasa AL, MacAvoy MG, Ding YS, Gelernter J, Carson RE. Amyloid-s burden and neuropsychological test performance in cognitively normal first-degree relatives at varying genetic risk for Alzheimer's disease. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Marshall GA, Olson LE, Frey MT, Maye J, Rentz DM, Sperling RA, Johnson KA. Instrumental activities of daily living impairment is associated with increased amyloid burden. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Becker JT, Price J, Kingsley L, Minhas D, Teverovsky L, Rinaldo CR. PiB retention and neuropsychological test performance among cognitively normal men. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Shaffer JL, Petrella JR, Doraiswamy PM, Sheldon FC, MacCarthy MD, Calhoun VD. Predicting cognitive decline in MCI subjects using Fusion ICA Toolbox to combine multimodality biomarker data. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Rodrigue KM, Kennedy KM, Devous Sr. MD, Park DC. Beta-amyloid in healthy aging: regional distribution and cognitive consequences. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
England RL, Becker JA, Olson L, Frishe K, Frey MT, Maye J, Johnson KA, Sperling RA, Rentz DM. Predicting amyloid deposition using multiple neuropsychological measures. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Landau SM, Marks SM, Mormino EC, Rabinovici GD, Oh H, O'Neil JP, Wilson RS, Jagust WJ. Cognitive activity is associated with amyloid deposition in normal older individuals. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Sheldon FC, Petrella JR, Doraiswamy P. Voxel degree as a measure of functional connectivity changes in Alzheimer's disease. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Sepulcre J, Becker A, Hedden T, Sperling R, Buckner L, Johnson K. Hubs of atrophy and amyloid-s in AD and normal aging. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Wang L, Schultz A, Dickerson BC, Becker JA, Johnson KA, Sperling RA. Amyloid deposition disrupts local coupling of intrinsic brain activity in cognitively normal elderly. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Vemuri P, Wiste HJ, Knopman DS, Lowe V, Weiner M, Petersen RC, Jack Jr. CR. Effect of APOE on rate of change of PIB retention across the cognitive continuum. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Price JC, Weissfeld LA, Bi W, Cohen AD, Saxton JA, Snitz BE, Berginc M, DeKosky ST, Mathis CA, Klunk WE. Evaluation of longitudinal PiB data using linear mixed models. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Ossenkoppele R, Tolboom N, Foster-Dingley JC, Boellaard R, Yaqub M, Windhorst AD, van der Flier WM, Barkhof B, Lammertsma AA, van Berckel BNM. Time course of specific [11C]PIB and [18F]FDDNP binding: paired studies in patients with Alzheimer's disease, mild cognitive impairment and healthy controls. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Rinne JO, Suotunen T, Rummukainen J, Alafuzoff I, Savolainen S, Helin S, Seppala T, Herukka S-K, Koivisto A, Leinonen V. Brain [11C]-PiB uptake reflects beta-amyloid burden detected by immunohistochemistry in frontal cortex biopsy specimens. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Sur C, Hostetler E, Connolly BM, Miller PJ, O'Malley S, Chen T-B, Culberson C, Harrison S, Mulhearn J, Zeng Z. Pharmacological characterization of a novel fluorinated PET tracer for the detection of amyloid-s plaques. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Pontecorvo MJ, Joshi AD, Skovronsky DM, Mintun MA. Florbetapir F 18 and 11C-PIB PET imaging provide concordant measures of underlying brain amyloid pathology. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Ikonomovic MD, Abrahamson EE, Mathis CA, Debnath ML, Srinivasan S, Hamilton RL, Klunk WE. Comparison of [H-3]Flutemetamol and [H-3]PiB binding to cortical amyloid in brains from Alzheimer's disease and control subjects. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Duara R, Loewenstein DA, Shen Q, Barker W, Potter E, Varon D, Heurling K, Vandenberghe R, Buckley C. [18F]flutemetamol PET imaging and medial temporal atrophy measures in distinguishing aMCI and AD from elderly normals. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Cselenyi Z, Forsberg A, Jonhagen ME, Halldin C, Julin P, Schou M, Johnstrom P, Varnas K, Svensson S, Farde L. Head-to-head comparison of amyloid-specific PET radioligands [18F]AZD4694 and [11C]AZD2184. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Das BC, Das S, Elmaleh DR. Design and synthesis of potential retinoid based PET imaging agents to study Alzheimer's disease biology. Human Amyloid Imaging 2011 Meeting Abstrac. 2011 Jan 15. Abstract
Nabi HA, Sajjad M, Dhindsa S, Erb D, Wack D, Chaudhuri A, Dubey S, Wisniewski S, Dandona P. Quantitative measurement of B amyloid plaques with C-11 PIB positron emission tomography of hypogonadal men with type 2 diabetes. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
MacCarthy MD, Petrella JR, Sheldon FC, Shaffer JL, Doraiswamy PM, Calhoun VC. Multi-modality fusion of neuroimaging and genetic data in predicting abnormal cognitive decline in aging. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Kantarci K, Przybelski SA, Lowe V, Weigand SD, Senjem ML, Ivnik RJ, Roberts RO, Geda YE, Boeve BF, Jack Jr. CR. s-Amyloid deposition, 1H MRS metabolites and cognitive function in a population-based cohort of cognitively normal elderly. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Nyborg A, Moll J, Wegrzyn R, Havas D, Hutter-Paier B, Feuerstein G, Rudolph A. In vivo preclinical administration of a novel peptide based imaging agent for Alzheimer's disease. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Christian BT, Wooten DW, Hillmer AT, Tudorascu D, Murali D, Barnhart TE, Harding S, Asthana S, Sager MA, Johnson SC. The relation of serotonin 5-HT1A receptors and amyloid load in prodromal AD. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Carter SF, Scholl M, Almkvist O, Wall A, Engler H, Langstrom B, Nordberg A. Investigating astrocytosis with 11C-deuterium Deprenyl in mild cognitive impairment and mild Alzheimer's .A multi-tracer PET paradigm. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Sojkova J, Goh J, Beason-Held LL, Zhou Y, An Y, Kraut MA, Wong DF, Resnick SM. Increased s-amyloid deposition is related to regional cerebral blood flow in nondemented older adults. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Hedden T, Van Dijk K, Shire E, Sperling R, Johnson K, Buckner RL. Relationship of neuropathological markers of white matter burden and amyloid accumulation to attentional control during aging. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Cohen AD, Price JC, Rudolph M, Jones Z, Rosario BL, Weissfeld LA, Nebes RD, Saxton JA, Snitz BE, Klunk WE. Comparison of cerebral metabolism using the PALZ tool in clinically unimpaired elderly and MCI subjects with and without amyloid deposition. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Becker JA, Maye J, Gidicsin C, Rentz DR, Hedden T, Marshall G, Olson L, Buckner RL, Sperling RA, Johnson KA. FDG metabolism, amyloid deposition and APOE status in cognitively normal elderly subjects. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Rostomian A, Madison C, Rabinovici G, Jagust W. Early 11C-PIB frames and 18F-FDG measures are comparable. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Lopresti BJ, Klunk WE, Cohen A, Coleman R, Weissfeld L, Mathis CA, Price JC. Consideration of pons normalizing region for [11C]PIB PET scans. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Wack DS, Wisniewski S, Erb D, Dandona P, Nabi HA. Complex singular value decomposition improves the image quality of C11-PIB PET images. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Zubal G, Wisniewski G, Marek K, Seibyl J. Automated image quantification methods of PET betaamyloid scans. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Ng Y-B, Carter S, Scholl M, Kadir A, Nordberg A. Amyloid deposits in the cerebral cortex of patients with Alzheimer's disease align with cytoarchitectonic properties. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Thurfjell L, Lundqvist R, Lilja J, Vandenberghe R. A software application for automated analysis of [18F] flutemetamol amyloid imaging data. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Lowe VJ, Weigand SD, Jack Jr. CR, Kantarci K, Senjem ML, Petersen RC. Optimized ROI measures of FDG PET improve characterization of early cognitive impairment and are comparable to PIB PET. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Mathis CA, Holt DP, Huang GF, Lopresti BL, Debnath ML, Shao L, Klunk WE. Species-dependent metabolism of [C-11]PiB. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
Schmidt ME, Mortishire-Smith R, van Vlaslaer A, Christiaens T, Langlois X, Mackie C. Profiling of hepatic clearance pathways of PIB with cytochrome P450 phenotyping. Human Amyloid Imaging 2011 Meeting Abstracts. 2011 Jan 15. Abstract
This concludes a seven-part series of the HAI Amyloid Imaging conference. See also Part 1, Part 2, Part 3, Part 4, Part 5, and Part 6. View a PDF of the entire series.
No Available Comments
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.