CONFERENCE COVERAGE SERIES
International Conference on Alzheimer's & Parkinson's Diseases 2007
Salzburg, Austria
14 – 18 March 2007
CONFERENCE COVERAGE SERIES
Salzburg, Austria
14 – 18 March 2007
Every other year, Abraham Fisher of the Israel Institute for Biological Research in Ness-Ziona, and Israel Hanin of Loyola University in Maywood, Illinois, team up with two local scientists from a different European country to host an international meeting. This year, the hosts were Werner Poewe, who chairs the neurology department at the Medical University of Innsbruck, and Manfred Windisch, who heads the Austrian contract research organization JSW-Research Ltd. in Graz. The International Conference AD/PD has been growing steadily; this year it drew nearly 2,200 people, up from 1,500 two years ago in Sorrento (see ARF related news story). The flood of registrants—1,800 were expected—led to a last-minute scramble to build a lunch tent outside the conference center and to locate additional hotel rooms. The conference proceeded smoothly, yet its rapid growth prompted some soul-searching among the organizers about how to keep the program sharp and logistics efficient while also preserving the personal flair that has made exchanging ideas and striking up new acquaintances easy at this conference before. At the end of the meeting, the organizers invited ideas from the attendants on how to keep improving. Some mentioned increasing the number of younger and female speakers, as well as offering travel awards to students, but additional suggestions are welcome.
The conference began with a tribute Windisch paid to Leon Thal (see ARF related news story) as a clinician-researcher, friend, and fellow aviator. Then the Austrian Oleh Hornykiewicz, who conducted the first clinical experiments infusing levodopa into PD patients in the 1960s, took history buffs and people too young to remember back to this decade. He told the story of how difficult it had been in those days to convince his fellow brain anatomists that an important projection from the substantia nigra to the striatum existed in the first place.
The conference offered no major news breakthroughs but many tidbits on topics spanning the complex spectrum of age-related dementias and movement disorders, of which Alzheimer's and Parkinson's are merely the best-known ones. One trend apparent at the conference was a vigorous effort to delineate forms of frontotemporal dementia anew in light of the discoveries last year of mutations in the progranulin gene and the identification of the TDP-43 protein as a major component of characteristic intranuclear inclusions. This gives researchers new tools to make sense of the overlapping clinical pictures, genetics, and pathologies of diseases ranging from different dementias all the way to amyotrophic lateral sclerosis. It also spurred a burst of studies to explore how these players fit into the disease pathogenesis and whether they could become therapeutic targets, though this research is only just gearing up and has few results to offer yet. Of several new mouse models introduced at the conference, one in particular illustrated this crossover between diseases, when a tau transgene from a family with the frontotemporal dementia Pick disease, expressed in mouse striatum, gave rise to a rather classic phenotype of parkinsonism and even responded to the PD medicine L-dopa.
In mouse model research more generally, a current trend in the field is to improve them by recreating various sorts of "multiple hit scenarios." Oxidative stress and mitochondrial impairment are typically a general component of this scenario and, combined with disease-specific transgenes, make neurons particularly vulnerable. These experiments either take the form of cross-breeding mouse strains or manipulating neurons cultured from transgenic models. Other translational research in mice strengthened the promise of ACAT inhibition for the future treatment of AD.
Another trend strengthened at the meeting was that efforts to test and validate preclinical biomarkers are beginning to dovetail in that CSF and imaging data are beginning to overlap into signatures that predict who will go on to develop full-blown AD from among a diverse group of people with mild cognitive impairment, and even cognitively normal people in some studies. Scientists are pushing back toward trying to predict AD reliably at a preclinical stage. Many talks throughout the conference echoed what is emerging as a consensus theme, namely that AD, and probably also PD, develop silently during a so-called prodromal phase, and that it is this phase during which therapeutics stand the best chance of working.
In Parkinson disease, efforts to develop antecedent markers are coming to the fore, both in CSF and serum, and even with techniques such as ultrasound. In AD, such work has been going on for a longer period of time. In imaging and biomarker research, efforts at coordinating some of the individual groups' work into national efforts are being discussed in various countries. For example, Japan, Germany, and Australia are starting up projects similar to the U.S./Canadian Alzheimer Disease Neuroimaging Initiative (ADNI), and internal discussions are underway to try to make the databases interoperable so that they support analysis of much larger datasets than any individual center, or even country, can generate.
Talks on immunotherapy offered little hard news, but many speakers converged on the theme that it should ideally become a preventive therapy eventually, given during the prodromal phase before plaques have built up. Alternatively, researchers agreed, it will have to be adjusted carefully so as not to overwhelm the capacity of the AD brain to clear away a wave of antibody-protein complexes washing up against aging blood vessel walls. One point of debate in immunotherapy research revolves around the relative values of N-terminal versus C-terminal antibodies, and active versus passive regimes. But several scientists pointed out that these kinds of categorical distinctions may be less important in determining which antibody is safe than the specific idiosyncratic properties of a given antibody. One open question at this time surrounds the significance of the fluid-attenuated inversion recovery (FLAIR) MRI images that were seen in the phase 1 and the currently ongoing phase 2 trial of Bapineuzumab, aka AAB 001. Such signals generally are seen as indicators of small hemorrhages. As always when there is no definitive data yet, speculation fills the void, and at the conference, opinions on the FLAIR signal ran the gamut from "passive immunization is finished, clinicians won’t risk causing bleeds" to "this is clinically irrelevant; our patients who show FLAIR signals are doing fine." Further research is needed to sort out this issue.
There is much activity on γ-secretase inhibitors and modulators. Companies presented ongoing programs, suggesting that industry has not abandoned γ-secretase as a major target following the discovery of mechanism-based side effects on Notch signaling, but is instead working to circumvent this problem. Scientists from several companies speculated that even if the first suggested γ-secretase modulator already in phase 3 trials, Flurizan (see ARF related news story), obtains FDA approval, its clinical benefit will be small enough to leave room for improvement. BACE secretase inhibitors are inching their way toward early clinical tests. A physiological role for BACE in the myelination of axons has pointed out an area to watch for potential side effects, and a growing number of research laboratories are exploring feverishly what regulates BACE. Other genes, proteins, inhibitory RNAs, environmental influences—all are under the microscope in the search for understanding new BACE-related targets.
On the therapy front, clinical trial results for a few agents were presented, but none surprised with strong effects just yet. The frontrunner of anti-amyloid therapies, Neurochem's Alzhemed, has completed its North American phase 3 trial. Data are being analyzed this month and will be presented in June at the Alzheimer's Association International Conference on Prevention of Dementia (see ARF related conference story) in Washington, D.C., according to Neurochem representatives. The makers of anti-amyloid drugs are vying to persuade the FDA to allow them to claim on their drug's label that it modifies disease rather than just treats its symptoms, and several presentations dealt with how clinical trial design would have to change to make this possible. While companies are wrestling with the difficult scientific problems posed by a new generation of experimental anti-amyloid therapies, a number of more traditional transmitter-based approaches were presented. They include serotonin receptor antagonists, butyryl receptor inhibitors, α7 nicotinic receptor agonists, and even estrogen receptor β agonists.
Approaches that have failed in the past, such as gene therapy and agonists of M1 muscarinic acetylcholine receptors, are making a careful comeback. An M1 agonist originally developed by Abraham Fisher has passed two single-dose phase 1 trials without incident and is about to enter a first multi-dose phase 1 study. Phenserine (see ARF related news story), a cholinesterase inhibitor that is thought to also affect amyloid levels, impressed some colleagues when Swedish scientists reported that it not only improved cognition and clinical symptoms somewhat in people with AD but also lowered their brain amyloid load as measured by PIB PET and shifted their CSF amyloid markers, as well. The reassurance came not so much from the drug’s small effect on cognition, but from its concurrent nudging of two separate biomarkers. Last but not least, the much-dismissed notion of a role for infectious organisms in neurodegenerative diseases is tenaciously hanging on, this time with anecdotal reports that treating Helicobacter pylorus infection in Parkinson patients who also have gastritis rather startlingly improved their PD. On the inflammation front, microglial imaging and experimental uses of the antibiotic minocyline to reduce inflammation drew note. Stay tuned for more detailed stories from the conference.—Gabrielle Strobel.
The science of frontotemporal dementias generated perhaps the loudest buzz at the 8th International Conference AD/PD 2007, held 14-18 March in Salzburg, Austria. The excitement grew out of last year’s twin discoveries that mutations in the progranulin gene account for a large chunk of heretofore unexplained cases of this diverse group of diseases (Baker et al., 2006; Cruts et al., 2006; Mukherjee et al., 2006) and that the protein TDP-43 is the main constituent of their characteristic misfolded protein inclusions (see Neumann et al., 2006; Arai et al., 2006). These were major findings that gave impetus to neurogeneticists, neuropathologists, and molecular neurobiologists around the world. Follow-on studies are gearing up, and big discoveries on how those proteins function and how they interact in the chain of pathogenic events are not in yet. But already, six papers on TDP-43 and 19 papers on progranulin were published on the heels of the initial reports, and the Salzburg conference featured nearly 30 presentations on the two proteins. Together, this work is beginning to paint a picture of these formerly obscure forms of neurodegeneration.
As an observer, this reporter was struck that the results coming in from several different groups all seem to jibe rather well, with few contradictory data sets as yet. A trend afoot in this field appears to be that the findings on progranulin and TDP-43 are prompting a realignment of the overlap generated by the different ways to characterize these diseases, i.e., the clinical description, the genetics, and the pathology. Finally, this growing field reminds us yet again that scientists and clinicians have to come to grips with the complex reality of spectrum disorders. Neurodegenerative disease occurs on continuums, not in the neatly defined boxes that an ordering mind tends to prefer. Below is a fairly comprehensive summary of new developments in Salzburg.
In her plenary lecture on TDP-43, Virginia Lee from the University of Pennsylvania School of Medicine, Philadelphia, noted that she expects this protein to define a new type of neuropathology with a similarly broad sweep as tau and α-synuclein have done before it. These better-known proteins first explained pathology, then opened a window on pathogenesis, and then became drug targets. TDP-43 may follow the same path, Lee said. Frontotemporal dementias (FTDs) were recognized in the mid-1980s. They account for close to half of all cases of dementia in people younger than 65. FTDs are distinct from Alzheimer disease, though differential diagnosis frequently is a problem. FTDs can be dramatic for families in that in addition to functional deficits, patients exhibit bizarre personality changes and psychiatric symptoms that are difficult to cope with. FTDs have a strong familial component. Their genetics first broke with the discovery in the early 1990s of mutations in the tau gene (causing FDTP-17), and since then some additional cases were pinned on mutations in the gene for valosin-containing protein, the CHMP2 gene, and an unknown gene on chromosome 9 (see AD/FTD mutation database). But those latter gene changes are rare, and part of the reason why progranulin energized scientists last summer is that it explains a large fraction of the remaining genetic burden of familial FTDs.
TDP-43 did for pathology what progranulin did for genetics. Its discovery suddenly accounted for the largest fraction of previously mysterious protein deposits in all FTDs. These are the ubiquitin-positive but tau-negative intranuclear, cytoplasmic, and neuritic inclusions that mark the brains of people with what was accordingly called FTLD-U. FTLD-U is the most common form of the FTDs. TDP-43 pathology occurs in a spectrum of people who have FTLD-U, FTLD-U with motor neuron disease, and even in sporadic cases of ALS. Sometimes, a given family will have one affected member suffering from frontotemporal dementia and another from ALS, but both people will have TDP-43 deposits in their respective sets of degenerating neurons. Taken together, the cytoplasmic and intranuclear inclusions seen across many different neurodegenerative diseases now fall into four main categories, Lee said. They are the
The finding that TDP-43 constitutes the main ingredient of ubiquitin-positive inclusions in the brains of people with progranulin mutations gratified researchers. As was the case with amyloid-β and tau before, a convergence of genetics and pathology always gives science a boost. But the connection between progranulin and TDP-43 is not exclusive. TDP-43 is broader. It also deposits in FTLD-U cases with mutations in the gene for valosin-containing protein, in FTLD-U cases without mutations in the known genes (Seelaar et al., 2007; Neumann et al., 2007), as well as in familial ALS without SOD1 mutations (Tan et al., 2007). And, dropping a small fly into the ointment for those who like to keep things nicely separated, Dennis Dickson of the Mayo Clinic in Jacksonville, Florida, reported in Salzburg that TDP-43 inclusions also occur in a subset of cases with AD. As a take-home message from the current pathology data, Lee suggested that TDP-43 defines FTLD-U and ALS as syndromic variants of the same pathology (also Davidson et al., 2007). That is, TDP-43 deposits in cortex and hippocampus underlie symptoms of FTD; in spinal cord and motoneurons they give rise to symptoms of ALS; and a mixed pathology gives rise to a mixed clinical phenotype in the middle of this spectrum. Moreover, both neurons and glia express TDP-43, and indeed the protein reveals a prominent, previously underappreciated white matter pathology in the cortex, spinal cord, and brain stem of people with FTLD-U/ALS (Neumann et al., 2007). TDP-43 constitutes a robust pathologic marker, and smart neurochemists may soon be trying to develop a brain imaging tracer for it.
TDP-43 is a conserved nuclear RNA-binding protein thought to be involved in transcriptional repression and in scaffolding structures called nuclear bodies. In deposits, TDP-43 is hyperphosphorylated, ubiquitinated, and truncated into C-terminal fragments. The disease process by which it becomes adulterated in this way remains unclear, nor is it known which genes it normally represses. TDP-43 gets drawn into a pathogenic process, partly by not being degraded properly anymore. However, it does not at present appear to be a major genetic cause of neurodegeneration. In Salzburg, neither a Belgian team led by Marc Cruts and Christine van Broeckhoven at the VIB-University of Antwerp, nor a U.S.-Canadian team led by Roos Rademakers and Mike Hutton at the Mayo Clinic in Jacksonville, Florida, reported finding disease-causing TDP-43 mutations in FTLD-U/FTD/FTD-ALS cases analyzed so far. Sequencing and association studies are ongoing, so a small genetic role for TDP-43 may still turn up. Progranulin, by contrast, is fast becoming a heavy-hitter in FTD genetics. The relationship between TDP-43 and progranulin is a mystery investigators would love to solve. For more on progranulin, see Part 2 of this meeting report.—Gabrielle Strobel.
No Available Further Reading
The 8th International Conference AD/PD 2007, held 14-18 March in Salzburg, Austria, offered a good opportunity for scientists at large to get up to speed on progranulin, the growth factor whose gene on chromosome 17 is quickly proving to be a major cause of frontotemporal dementia. Since the discovery last summer that mutations in this gene account for many cases of FTLD-U marked by tau-negative but ubiquitin-positive protein inclusions, scientists all over the world have jumped at the chance to understand progranulin better. In Salzburg, the news came in three flavors: There were clinico-pathological observations, genetics, and a budding interest in how a loss of progranulin might cause neurodegeneration. First, the clinical observations.
Several different research groups described their patients with progranulin mutations. The general tenor of these talks and posters was that from a pathological perspective, the patients all had similar inclusions containing ubiquitinated TDP-43. Brain imaging always revealed an asymmetrical pattern, with atrophy and hypoperfusion in certain areas of only one side of the patient’s frontal lobes, indicating that one side of the brain degenerates selectively. And yet, despite these commonalities, these patients came to see their doctors with a surprisingly wide range of complaints that gave rise to an equally wide range of clinical diagnoses. Often there was a spectrum in a given family, and often a person’s diagnosis changed in the course of his or her illness as additional symptoms emerged.
Consider some examples. B.J. Kelley from the Mayo Clinic in Rochester, Minnesota, recounted the story of a family with eight affected members in three generations. One had a dementia diagnosis; two had an Alzheimer or first-MCI-then-AD diagnosis, one had an AD diagnosis that was later changed to FTD. In the third generation, one person is living with amnestic MCI, one received a diagnosis of primary progressive aphasia (PPA) that progressed to PPA/FTD, and one had a diagnosis of FTD. In this family, the affected members all had memory impairment in addition to their other symptoms. In a second family, the diagnoses ranged from AD to behavioral features more typical of FTD, and to behavioral features followed eventually by a Parkinson’s diagnosis. In a third family, the first case first had symptoms of idiopathic Parkinson’s and later was diagnosed as having Parkinson’s with dementia. A sibling had FTD, another had FTD with parkinsonism, and a third had parkinsonism, dementia, and disinhibition and personality changes typical of FTD. All told, Kelley noted that 31 people with a wide range of clinical problems all turned out to have a progranulin mutation, and they also all had the FTLD-U pathology.
Michael Hutton, of the Mayo Clinic in Jacksonville, Florida, said that the most frequent clinical diagnoses people with progranulin mutations receive are for frontotemporal dementia and primary progressive aphasia (PPA); indeed, language difficulties are often what prompt the patient/family to come to the clinic. Corticobasal syndrome also can be an expression of progranulin mutations, a different Canadian/U.S. team reported. Of the patients that Hutton’s group follows, the mean age at death was 63. The mean age of onset was 59, but as with familial AD, the onset range spans some 30 years. Curiously, several scientists noted that later generations in an affected family may show onset at younger ages than their parents, as if they anticipated disease in themselves. This is distinct, however, from triplet repeat diseases, where the abnormal DNA repeats are known to grow in length from one generation to the next, driving the onset age down. The penetrance, that is, a person’s chance of getting sick, also increases with age, Hutton noted. By age 60, about half of all progranulin mutation carriers are ill, and nine of 10 are by age 70.
Ian Mckenzie of the University of British Columbia in Vancouver, Canada, sounded a similar note. Mackenzie emphasized how the characteristic pathology of people with progranulin mutations unites with striking consistency a broad swath of clinical presentations. Last November, Mckenzie and colleagues published the neuropathological features of 13 of their cases (Mackenzie et al., 2006) and in Salzburg he presented an expanded version of that data. In summary, his group, too, found atrophy of the frontal lobes, as well as degeneration of the substantia nigra and medial thalamic nuclei. Mckenzie described abundant, lens-shaped neuronal inclusions in the neocortex, striatum, and hippocampus that stain with antibodies against ubiquitin and TDP-43. The hallmark lesions are inclusions in the nuclei of neurons, but the cytoplasm, some neurites, and some glia have them, as well. The inclusions do not contain progranulin protein and are granular, not filamentous, a finding Virginia Lee’s group, too, saw by electron microscopy. FTLD-U cases without a progranulin mutation had a similar but milder pathology, Mckenzie said.
Samir Kumar-Singh and colleagues from the VIB-University of Antwerp presented pathological data of the Belgian founder family carrying a progranulin-null mutation, described in Cruts et al., 2006. Describing six patients from this extended family, Kumar-Singh emphasized that he observed pathological heterogeneity in the predominant lobar atrophy (parietal vs. frontal lobe atrophy) and also in the subcellular localization of the inclusions (nuclear vs. cytoplasmic) or pathology within neurites. Again, all these ubiquitinated deposits were tied together by the presence of TDP-43 protein. While TDP-43 is always there, it is not the only deposited protein in the ubiquitinated inclusions. Cruts noted that other known proteins were also abundant within them, notably p62.
On the genetics front, the data is branching out from the initial null alleles published last summer by the international teams of Hutton, Howard Feldman at the University of British Columbia, Vancouver, Canada, and Christine Van Broeckhoven at the VIB-University of Antwerp, Belgium. Thirty-nine different mutations in more than 70 families have been found so far, Hutton said, all of which cause a loss of protein function. The scientists traced the most frequent progranulin mutation, R493X, back to a founder effect in England, and were able to follow its distribution to the countries of English emigration, such as the U.S. and Australia. To date, progranulin mutations in Hutton’s samples explain roughly 5 percent of frontotemporal dementia and 13 percent of cases with a family history; this compares with 9 percent and 22 percent, respectively, in a French-Belgian case series led by Alexis Brice at INSERM in Paris and Van Broeckhoven. As always in such studies, occasional cases thought to be sporadic also turn out upon sequencing to have previously discovered familial mutations. A study by Stuart Pickering-Brown from Manchester University estimated the genetic contribution of progranulin to FTD to equal that of tau in a British series of 270 cases, while in a Belgian series, progranulin’s contribution is larger than tau’s. Van Broeckhoven’s team added progranulin genomic deletions to the spectrum of null mutations that may explain an additional 2 percent of FTD. By contrast, researchers led by Lena Skoglund at Uppsala University in Sweden reported that initial analysis of Scandinavian FTD patients turned up little, suggesting progranulin is not a major cause of FTD in Europe’s northern realms.
Other presentations in Salzburg reflected geneticists’ ongoing search for other types of genetic flaws that would reduce a person’s progranulin protein levels beyond outright null alleles, such as missense mutations leading to dysfunctional protein or promoter mutations that depress transcription. All Belgian studies mentioned below came from Van Broeckhoven’s group. One Belgian-French collaboration reported finding three new missense and three promoter variants after direct sequencing of the progranulin gene in 332 FTD patients from these two countries (see van der Zee et al., 2007). Scientists are also exploring the genetic contribution of progranulin to diseases other than FTLD-U. A different sequencing study in a Belgian sample of 779 clinical AD cases turned up new progranulin missense mutations in addition to finding some of the original progranulin-null mutations linked to FTLD-U in a Belgian founder family. Besides further reinforcing the clinical heterogeneity of progranulin mutations, this finding raises the possibility that the gene might end up accounting for a small fraction of AD cases, as well. A separate Belgian study sequencing progranulin in 270 clinical PD cases produced fewer hits, suggesting that progranulin mutations are not a major cause of PD but should be considered in cases where PD patients show simultaneous clinical symptoms of dementia. The role of progranulin mutations in ALS is in flux. Van Broeckhoven noted that sequencing the gene in 230 Belgian ALS patients turned up 11 progranulin mutations, some of which were predicted to affect progranulin protein sequence or levels, plus a common variant that appeared to drive down age of onset and make the disease more aggressive. By contrast, last month a U.S. consortium of scientists concluded that progranulin mutations are not a common cause of ALS in 361 cases of either sporadic or familial ALS, or ALS-FTD (Schymick et al., 2007).
All these different mutations boil down to lowering the amount of progranulin protein in the brain. How does too little of this secreted growth factor cause disease? In addressing this open question, Hutton laid out opportunities for mechanistic and, eventually, therapeutic studies (see also Ahmed et al., 2007). Both neurons and microglia express progranulin, while astrocytes and oligodendrocytes typically do not. Any injurious stimulus that activates microglia will induce a dramatic increase in progranulin expression, Hutton noted. This happens in a host of diseases, and also in mouse models of neurodegeneration. For example, P301L tau transgenic mice massively overexpress progranulin compared with wild-type mice by 12 months of age, and this goes along with activated microglial staining and tau pathology. Aging itself also increases progranulin expression, but less so. In AD brain as well as in transgenic mouse models, progranulin in microglia accumulates in large amounts around amyloid plaques.
Progranulin comprises seven conserved repeats, which, when cleaved by the enzyme elastase, can release smaller peptides called granulins. Progranulin is known to play an important role in tissue repair. It appears as if the whole protein and its smaller granulin offspring have opposing roles in regulating the inflammation that can accompany wound healing. Why is progranulin present in such high levels in activated microglia? Its established role in peripheral wound healing might point to the answer of this question, Hutton noted.
In the periphery, progranulin stimulates the growth of epithelial cells close to the wound, and the site of injury becomes bathed in the growth factor. With wound repair typically comes an increase in inflammatory cytokines, and the conversion from progranulin to individual granulins serves as a molecular switch to control inflammation. If a similar regulatory system operated in the brain, one could imagine that progranulin released from neurons normally is neurotrophic, but in an inflamed or stressed situation, elastase-generated granulins become phagocytotic. This idea is currently under study.
Hutton noted that the clear-cut impact of the various known mutations had his group excited because it implied that restoring progranulin levels might be therapeutic. One way in which that appears doable from an industrial drug development perspective would be to find a small-molecule drug that boosts expression of the remaining normal progranulin gene. After all, the mutation is heterozygous. Initial studies surprised Hutton’s group when a large number of compounds from well-characterized chemical groups turned out to be able to do just that. For example, ibuprofen and other NSAIDs increase progranulin production in cells, Hutton said. In theory, if one can safely induce sustained progranulin elevation in brain, then that could potentially prevent disease in patients who have mutations. Much work remains to be done on this concept. For now scientists are enjoying the early days, when the limited knowledge on progranulin still fits into a simple picture, and the proverbial devil in the detail has not yet reared its ugly head.—Gabrielle Strobel.
No one disputes that Alzheimer’s, as well as other neurodegenerative illnesses, are in part diseases of misfolded protein piling up in neurons. Yet it has been difficult to study exactly where things go awry because some of the parts of the molecular machinery with which neurons dispose of misshapen proteins have been mysterious. At the 8th International Conference AD/PD, held last month in Salzburg, Austria, Mervyn Monteiro of the University of Maryland Biotechnology Institute in Baltimore offered up a new protein complex for the field to study. Monteiro suggested that the new protein erasin forms a platform on which the proteins p97 (aka VCP) and ubiquilin settle down as they yank potential troublemakers out of the lumen of the ER and render them to the proteasome for degradation.
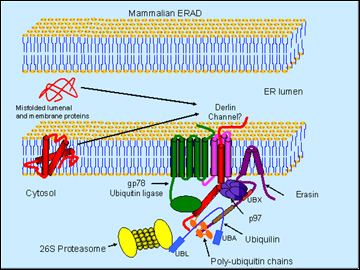
Schematic model of erasin-p97-ubiquilin complex in ERAD (Image credit: Mervyn J. Monteiro)
This proposed garbage crew helps shed new light on a regulated process called ERAD, short for endoplasmic reticulum-associated degradation. ERAD has its own literature going back a decade, but researchers in neurodegenerative diseases have not been able to get a good fix on its molecular players. Monteiro’s new science is of the basic kind, though disease connections for the involved proteins abound. P97 is none other than the valosin-containing protein known to cause rare familial of frontotemporal dementia with ubiquitin and TDP-43-positive inclusions (see ARF Salzburg story), as well as inclusion body myopathy and Paget's disease of bone. Ubiquilin is a presenilin-binding protein found in postmortem tissue of AD brain and in huntingtin aggregates. In research models of triplet-repeat diseases, ubiquilin protects against polyQ-expanded huntingtin and the formation of polyQ inclusions. It is one of many genes for which some genetic association data exist, though as with many candidate genes, other studies to date have not reproduced initial findings (see Alzgene). More detailed study of these proteins may shed light on the common aspects that many protein misfolding diseases share.
Monteiro presented data to suggest that manipulating poly-Q huntingtin in the worm C. elegans supported the idea that it facilitates degradation of this misfolding protein. Ubiquilin overexpression decreased both protein aggregation and a movement phenotype, whereas RNAi knockdown exacerbated these readouts (Wang et al., 2006). Ubiquilin sped up the selective degradation of the expanded huntingtin. But how might it do that? To explain, Monteiro introduced erasin (Liang et al., 2006), an ERAD protein that interacts with ubiquilin. Thanks to a short hydrophobic region, erasin is firmly anchored in the ER while dangling most of its sequence into the cytoplasm, where it exposes two important binding sites. One of those, a UBX domain close to the membrane, binds the p97/VCP; the other one is a coiled-coil motif farther down the protein, which binds ubiquilin.
Here is Monteiro’s working model of this complex in a nutshell: erasin forms a platform that recruits p97/VCP and ubiquilin. The former is a molecular chaperone that extracts misfolded proteins from the ER, possibly through channels called derlins. The trash-to-be gets ubiquitinated by a ubiquitin ligase (such as gp78) as it enters the cytosol. Because the UBA domain of ubiquilin can bind poly-U chains and ubiquitin is positioned conveniently downstream of p97 on the erasin protein, the tagged protein gets handed on to ubiquilin. Then the N-terminal UBL domain of ubiquilin binds the proteasome and delivers the trash into its teeth.
As supporting evidence, Monteiro showed data suggesting that erasin levels shoot up with ER stress and that proteasome inhibition increases ubiquilin-erasin binding. In collaboration with Ralph Nixon at the Nathan Kline Institute in Orangeburg, New York, the scientists showed that erasin antibody staining is increased in AD postmortem tissue. Monteiro’s take-home message is that the complex of erasin, ubiquilin, and p97/VCP helps clear misfolded proteins from the ER.—Gabrielle Strobel.
No Available Comments
At the 8th International Conference AD/PD, held last month in Salzburg, Austria, Dora Kovacs of the Massachusetts General Hospital presented new data advancing her story of ACAT inhibition as a potential new therapeutic approach to Alzheimer disease. Following an earlier report that the pharmacological compound (read: not a real human drug) CP113,818 reduces amyloid deposition in transgenic mice (Hutter-Paier et al., 2004), many observers have been curious where this approach would go next. In Salzburg, Kovacs reported that the compound avasimibe (a real drug) performs almost as well, validating the general approach.
Avasimibe is a small-molecule inhibitor of the enzyme ACAT-1. It counteracts the formation of cholesteryl esters from free cholesterol and fatty acids. The esters accumulate in the foam cells seen in atherosclerosis. Avasimibe had made it to phase 3 trials for cardiovascular disease, but the pharmaceutical giant Pfizer discontinued its development a few years ago. Internal drug interaction research apparently showed that avasimibe weakened the effect of lipitor, Pfizer’s leading cardiovascular drug, when it had instead been meant to boost lipitor and be taken alongside it (see Forbes.com; Sahi et al., 2003). Avasimibe also underwhelmed in the clinic (Tardif et al., 2004), and onto the dung heap it went.
AD aficionados will know that none other than Ronald Reagan was famous for quipping: “There might be a pony in there” when faced with bad news. He might have invoked his joke here, too, for when Kovacs tested two doses of avasimibe in APP/PS transgenic mice and wild-type mice, she found that the drug favorably changed amyloid endpoints. Working with scientists at the Austrian CRO JSW Research in Graz, Kovacs found that two months of treatment with 14.4 mg/kg of avasimibe per day reduced both serum and brain cholesteryl ester levels in the mice. In six-month old mice, it also reduced the brain’s plaque load by two thirds. To better model AD, where patients already have high plaque loads before they begin therapy, the scientists treated four 14 month-old transgenic mice whose brains were laden with amyloid. Avasimibe reduced diffuse deposits labeled by the antibody 6E10 but not core plaques stained with thioflavin S. Kovacs noted that she interprets her data as suggesting that the ACAT inhibitor interferes with Aβ production, allowing endogenous clearance mechanisms to remove diffuse deposits and reduce the net burden of Aβ. Kovacs’ lab attacked ACAT-1 by a third method, this time using siRNA in cultured cells. This, too, reduced total Aβ, Aβ42, and APP CTF generation. These experiments showed that a modest halving of ACAT-1 protein was sufficient to drive down cholesteryl ester levels by a fifth and Aβ secretion by 40 percent (Huttunen et al., 2007).
The gene for ACAT currently occupies rank 12 on the list of Top Alzgene Results, mainly thanks to a study that reported a common protective polymorphism associated with low brain amyloid load (Wollmer et al., 2003). How might it work? To get at the mechanistic underpinning of APP processing by ACAT-1, Kovacs, Henri Huttunen, and colleagues used the inhibitors as tools to find proteins that interact with APP in a way that is sensitive to ACAT inhibition. Once explored more deeply, this approach could reveal new pathways of APP processing. In Salzburg, Kovacs reported initial data that the ER chaperone GRP94, and the chaperone/protease HtrA2/Omi appear to bind APP in this way. Both proteins respond to ER stress, and both normally function somehow in the maturation and degradation of APP in the ER. While HtrA2 is primarily known as a mitochondrial protein, Kovacs and colleagues showed in Salzburg that it also occurs on the cytosolic side of the ER membrane and participates in ERAD (see related Salzburg story). This work is pointing to the endoplasmic reticulum as the cellular site at which ACAT inhibitors can influence the fate of APP.—Gabrielle Strobel.
No Available Comments
18 April 2007. Five years ago this spring, the molecular amyloid imaging probe PIB for the first time entered the veins and brains of people at the first independent site outside of the developer’s home turf at the University of Pittsburgh. The volunteers were patients at Agneta Nordberg’s outpatient clinic at the Karolinska University Hospital Huddinge in Stockholm, and they submitted to the first European test of this substance at the Uppsala PET center/Imanet in Uppsala, Sweden. At the 8th International Conference AD/PD, held last month in Salzburg, Austria, Nordberg recalled that right from the get-go in 2002, it was clear to the Swedish investigators that there was a stark difference in PIB uptake between people with and without AD. Even so, back then discussing amyloid imaging was a challenge and met much skepticism, said Nordberg, who is also at the Karolinska Institute. That has changed thoroughly since then, as some 25 groups around the world have begun their own independent studies to verify claims made for PIB, and to explore amyloid imaging more broadly and deeply. At this point, Nordberg said, the field can conclude that amyloid imaging works. It is also clear that amyloid PET imaging is not a great burden. Procedures vary slightly among the different groups, but typically a person receives a PIB injection into an arm vein while lying in the scanner, and is then scanned for 60 to 90 minutes. It is, however, still expensive. Several companies have jumped in to develop various ligands, and the scientific debate has moved on to subtler questions. This story summarizes some of the 15 presentations on amyloid imaging at the AD/PD conference. The talks and posters offered news morsels on the following issues:
First, the emerging consensus. Worldwide, more than 500 patients have been scanned with PIB to date, 200 of them in Australia alone. Several presenters noted that basic observations at the different centers in large part converge. For example, PIB reliably binds to β amyloid in the nanomolar range, and it visualizes amyloid deposits in a characteristic pattern of brain areas in different people with AD (see, e.g., Ng et al., 2007). Importantly, PIB imaging in the hands of several different groups indicates that a person’s brain amyloid load has built up more or less fully before the person is diagnosed with AD. The longest-standing studies are now beginning to report repeat scans of people who have progressed in their AD. They are finding that while the patients over time decline clinically, and also decline in terms of brain atrophy as measured by MRI and glucose utilization as measured by positron emission tomography (FDG PET), their PIB amyloid load stays unchanged (e.g. see Engler et al., 2006). This was surprising, as many colleagues had expected a linear PIB increase with ongoing disease. “The amyloid just seems to sit there stably as the patient gets worse,” said David Brooks of Imperial College, London. It could be that there is a steady state with active amyloid turnover that PIB imaging cannot detect, but the patient’s total value of PIB imaging tends to stay the same even years into the disease. This means that PIB is not going to be a marker for disease progression, and indeed several presenters noted that PIB load does not correlate with cognitive symptoms in clinically diagnosed AD. At the same time, this also suggests that PIB or other amyloid imaging ligands could be a useful marker for the effect of amyloid-removing therapies. This is a helpful refinement of what PIB can and cannot do, as a candidate biomarker typically cannot serve all of the potential purposes scientists initially envision for it—that is, marker for prediction, diagnosis, progression, treatment effect (see ARF related news story).
Early Detection
The realization that a person’s amyloid load has built up fully by the time of diagnosis comes only partly from repeat scans of AD patients who are being followed over time. It comes also from scans of people with MCI. In Salzburg, several groups reported data similar to what the Pittsburgh researchers have seen. Brooks’s Imanet center in London, Nordberg’s Swedish group, as well as Christopher Rowe’s group at Austin Hospital near Melbourne, Australia, and a fourth group led by Riitta Parkkola at the University of Turku’s Pet Center in Finland, all showed that about 60 percent of their MCI cases scanned with PIB so far (24 in London, 21 in Stockholm, 40 in Melbourne, 13 in Turku) have a PIB reading like that of AD cases. “My guess is these are the people who will get AD,” said Brooks, who also works for G.E. Healthcare, which develops PIB commercially. Groups around the world are following MCI cohorts to answer this question. Rowe noted that during the MCI stage, the PIB load correlates strongly with impaired cognitive function, and that this correlation only disappears once dementia is established as AD. In the Australian healthy aging group, people with high PIB readouts had slightly lower episodic memory performance and were frequently judged to be "declining," whereas people with low PIB readouts were judged to be “stable.” Rowe cautioned that this is an early observation based on too few patients to be statistically significant. Nordberg reported that in her group of 21 longitudinally followed MCI patients, 11 had high PIB and seven of them have since converted to AD. Of the people who had low PIB, none have converted (Forsberg et al., in press at Neurobiology of Aging). All three speakers noted that they expect amyloid imaging to help them with the challenge of predicting who will go on to develop AD from among the heterogeneous group of people who are classified as having MCI.
At the research level, the effort to predict future AD reaches even further back into groups of cognitively normal volunteers. The Australian group reported in Salzburg that about 20-25 percent of healthy people around age 70-75 begin to show significant amyloid loads in PIB PET scans. This matches postmortem findings. “It is tantalizing to speculate that these are the people who will have AD by age 85,” said Rowe. Other investigators, too, are asking themselves whether they are looking, literally, at the prodromal stage of AD in these people. Nordberg said that on this issue, the field would do well to explore more deeply the emerging correlations between PIB data and cerebrospinal fluid biochemistry of Aβ and tau proteins. She noted that Swedish data linking PIB and CSF appear to strengthen the prediction along the same lines as reported recently by researchers at Washington University, St. Louis, Missouri (Fagan et al., 2007). In addition, the significant percentage of high PIB levels in cognitively normal people implies that clinical studies must take care not to recruit people with subjective memory complaints or elevated AD risk factors into “healthy control” groups to avoid blurring the necessary distinctions between the trial groups, Nordberg added.
Budding Drug Marker?
Can amyloid imaging report on the success or failure of anti-amyloid drugs? This research is in its infancy, but Nordberg reported a first glimpse in Salzburg. A 1-year placebo-controlled Swedish trial of phenserine in 20 patients with mild AD incorporated PIB PET scans along with FDG PET, CSF and plasma biochemistry, and cognitive readouts. Phenserine is an experimental acetylcholinesterase inhibitor reported to modulate Aβ production. Between baseline and the 6-month time point, the scientists noted an improvement in MMSE and in an attention test in the patients on study drug. Some of the other markers also changed in these people: glucose metabolism increased, PIB retention nudged downward by about 12 percent (test-retest variation of PIB is below 5 percent), and CSF Aβ nudged upward. CSF tau stayed unchanged. “This is the first time we have related PIB measurements and CSF to a drug effect. The people on phenserine did slightly better and their biomarkers are moving, though we don’t understand it fully yet,” said Nordberg. For news on differential diagnosis, PIB biology, other amyloid imaging ligands, and imaging inflammation, see Part 2 of this story.—Gabrielle Strobel.
No Available Comments
Differential diagnosis clinicians face a daily challenge in properly distinguishing AD from related neurodegenerative diseases that can present a similar set of symptoms. Could amyloid imaging help? Presentations in Salzburg converged on the take-home message that PIB separates AD from Parkinson’s with later dementia and from frontotemporal dementia (FTD), but does not separate AD from dementia with Lewy bodies (DLB), and maybe not from cerebral amyloid angiopathy (CAA), either.
David Brooks’s London team reported two small series of 13 cases each. One showed that most people who develop Parkinson disease first and dementia later (i.e., PDD) did not show labeling with PIB even though their glucose utilization dropped. This suggests that they have no Aβ amyloid and that their dementia may be a consequence of Lewy body pathology. The other series scanned people with DLB, who develop dementia alongside their PD symptoms. Most of them did show significant PIB labeling, if not as much as did people with AD, suggesting that they have two different pathologies in their brains. The anterior cingulate was one of the first brain areas to show amyloid in people with DLB. Christopher Rowe’s Melbourne group did not study PDD but reported, like Brooks, that patients with DLB have amyloid deposition similar to AD patients. Rowe showed that all his cases with FTD looked like normal controls in PIB PET scans. Similarly, Timo Grimmer in Alexander Drzezga’s group at Technical University, Munich, Germany, described a study of semantic dementia (SD). This form of temporal lobe degeneration tends to strike at earlier ages than AD and is marked by deficits in word memory and comprehension. The clinical symptoms and the pattern of glucose metabolism with FDG PET of semantic dementia somewhat resemble those of AD. But at postmortem, the brains of people with semantic dementia show a characteristic pattern of neuronal loss without amyloid pathology, Grimmer noted. A comparison of eight SD and eight AD cases showed that the trained eye can distinguish them on FDG PET scans because the SD cases have reduced metabolism more in frontal and temporal cortex while AD cases have it prominently in temporal and parietal areas. But PIB PET distinguished much more clearly in that the SD cases showed no binding at all. (They did show a small amount of white matter binding, as do most PIB studies. That, however, reflects merely the tracer’s slower kinetics through white matter, not a specific binding to amyloid deposits, as presented on a poster by Michelle Teena Fodero-Tavoletti and other Australian researchers.) The German group suggested that semantic dementia arises from a pathology other than amyloid, and that amyloid imaging could help tell the two apart in cases where the diagnosis is in question.
Not Just Plaques: PIB Biology
The rush to explore PIB binding in people has left fundamental biologic research in its dust, said Andrew Lockhart of GlaxoSmithKline in Cambridge, U.K. The excitement about potential antecedent markers and drug effect monitoring has obscured the fact that the underlying biology and binding properties of PIB in tissue remain hazy (see Lockhart, 2006). PIB’s developers, Chet Mathis and William Klunk of the University of Pittsburgh, have conducted biochemical binding studies but have emphasized repeatedly that they do not know exactly which forms of Aβ amyloid PIB binds in people’s brains. This amyloid comes in many guises. Oligomers, diffuse deposits, neuritic plaque, cored plaque, parenchymal versus vascular amyloid—what exactly is behind those colored pictures?
Lockhart and colleagues set out to study the specificity of PIB binding to the major known types of amyloid lesion, and to other forms of non-amyloid deposits. To that end, he prepared a tritium-tagged version of PIB and treated fresh cryosections of human postmortem brain with it for quantitative autoradiography. The scientists used immunocytochemistry on adjacent sections for comparison. The study included sections from various forms of pathologically confirmed disease, including plaque-only AD, plaque-and-tangle AD, CAA, mixed parenchymal and vascular pathology, tangle-only or Lewy body-only cases of dementia. Lockhart found that PIB labeled all forms of amyloid plaque including diffuse plaques throughout all brain regions examined. PIB delineated the same structures that lit up with thioflavin S and the 6E10 antibody.
PIB did not bind to α-synuclein deposits, that is, Lewy bodies. This is consistent with Rowe’s and Brooks’s human imaging studies, in which PIB appeared to label only Aβ amyloid pathology in DLB patients who had a PIB-positive scan, and in which patients with PDD had PIB-negative scans. This implies that PIB will be useful for stratifying people with mixed-amyloid DLB from others with pure α-synuclein disease, Lockhart noted. Lockhart’s group found that PIB clearly delineated neurofibrillary tangles, but the labeling was much less intense than that of Aβ lesions, suggesting that in vivo PIB is essentially a specific reagent that measures total Aβ amyloid load.
Importantly, PIB also bound to vascular amyloid deposits, confirming a recent postmortem study (Bacskai et al., 2007). In a poster, Nordberg’s group also showed binding of PIB to blood vessels in brain tissue of people with CAA. Together, the findings imply that in vivo PIB PET images reflect Aβ-related amyloid but say little about the specific kind of amyloid people predominantly have in their brains, Lockhart said. One footnote: curiously, Lockhart found differences in PIB binding to cerebrovascular amyloid in the brains of ApoE4 carriers, an issue that needs more study, especially if PIB is to be used in monitoring preclinical AD in higher-risk people homozygous for ApoE4.
Beyond PIB: Other Ligands
PIB is the most thoroughly studied, but far from the only amyloid imaging probe. Others have entered the scene and are at various stages of exploration. In Salzburg, Rowe presented the first human data on a new ligand that has the great advantage in that it does not require an 11C radiolabel to make it “hot.” The short half-life of 11C requires a separate cyclotron run for each study, as well as specially trained chemists to couple the isotope to the ligand. By contrast, PET using the radioligand 18F is quite widely used in cancer imaging and would facilitate the spread of PIB past a small number of academic centers. Rowe’s center is testing the 18F AV-1/ZK ligand. It is related to the older stilbene SB13 ligand originally developed by Hank Kung at University of Pennsylvania, Philadelphia, and tested once in humans (Verhoeff et al., 2004). Avid Radiopharmaceuticals, also in Philadelphia, and Bayer Schering Pharma are developing the commercial product, and Rowe has an arrangement with them as a clinical investigator.
In Salzburg, Rowe presented first data on 10 AD patients and eight controls. Injected intravenously, the new probe caused no adverse events, and time-activity curves showed ready metabolism in both groups of people. The PET images with this new probe show the same distribution of label as PIB. Most controls did not retain PIB, and all people with AD had the highest binding in their precuneus, posterior cingulate, and frontal cortex, followed by lateral temporal and parietal cortex, as well as low binding in sensorimotor cortex. The non-specific white matter signal was also there. The caudate nucleus showed a little less binding than with PIB. At 3.03, the effect size of the AV-1/ZK is slightly smaller than PIB’s 3.7 but still represents a robust increase in AD over control, Rowe noted. Expressed in different terms, the relative increase in binding in AD of AV-1/ZK is in the range of 60 percent. This compares to 60-80 percent for PIB, and 10 percent for FDDNP, Rowe said.
Other compounds being developed include Japanese ones such as BF-227 (Kudo et al., 2007; Kudo, 2006). Preclinical MRI agents are promising (Higuchi et al., 2005). Several speakers in Salzburg mentioned the 18F compound FDDNP (Small et al., 2006), but the conference featured no presentations on it. Brooks noted that side-by-side comparisons in the same subjects of the different PET tracers would be interesting, but are complicated, thanks to licensing restrictions and academic rivalries.
Inflammation and Amyloid: See Both Together
The search for microglial tracers represents a new frontier in brain imaging, and the AD/PD conference offered some glimpses on how it might begin to dovetail with amyloid imaging. One such approach comes from Brooks’s attempts to visualize the inflammatory response to amyloid deposition with 11C-PK11195, an old compound that was developed originally at Parke-Davis as an anti-arthritis drug. It binds to microglia only when they are switched on. When an injury occurs in the brain (not just in AD), microglia swell, release cytokines, and express peripheral benzodiazepine binding sites that bind PK11195 (e.g., Tai et al., 2007). Brooks showed that wherever amyloid is deposited in AD cortex, his group sees activated microglia, that is, the distribution of the PIB and PK11195 corresponded closely. In PD, microglia are known to become activated, as well (Ouchi et al., 2005; Brooks, 2007). Brooks reported seeing a microglial signal come up not only in PD midbrain, but also throughout the brain in a distribution and strength that correlated with the Braak staging of Lewy body degeneration.
The question is whether activated microglia are helping to clear amyloid or are releasing damaging proteins, or both. Some drugs are known to switch off microglial signals, such as FK-506 (e.g., Yoshiyama et al., 2007), new PPARγ agonists, or minocycline. In the neurodegenerative disease multiple system atrophy, minocycline treatment tamps down the microglial signal, Brooks said. Both Siemens and GE Healthcare are developing newer microglial tracers than PK11195. Brooks hopes that the new compounds will soon put scientists in a position where they can try to treat brain inflammation pharmacologically, and accompany such a trial with both microglial and amyloid imaging as treatment-based biomarkers.—Gabrielle Strobel.
This is Part 2 of a two-part meeting report from the 8th International Conference AD/PD, held 14-18 March in Salzburg Austria. See Part 1.
 John Trojanowski
John Trojanowski
This is an important study. It needs to be extended to examining other types of brain amyloids (prion amyloid, alpha-synuclein amyloid, etc. and non-amyloid protein aggregates such as those formed by TDP-43 in FTLD) using similar methods and analytical techniques.
View all comments by John Trojanowski Hilkka Soininen
Hilkka Soininen
This paper reports an Aβ ligand (18)F-BAY94-9172 that, due to the half-life of (18)F, is suitable for clinical use. This is an important study and good news for clinicians.
View all comments by Hilkka SoininenWhile no one disputes that tau plays a critical role in diseases across a wide swath of the neurodegenerative spectrum, tau-based drug discovery efforts still lag behind those based around the amyloid hypothesis. This, researchers agree, is partly due to the difficulty of developing just the right mouse models, a problem that slows down mechanistic as well as therapeutic studies. Tau is catching up, though, and some of the energy was on display at the 8th International Conference AD/PD, held 14-18 March in Salzburg, Austria. This story offers some recent developments. It is not a comprehensive update, but as always, readers are welcome to improve it with their own observations from the meeting.
In reviewing mouse models for tau, Karen Duff of Columbia University, New York, noted that nearly two dozen have been made to date (see Alzforum Research Models), and these strains indeed model various aspects of tauopathy. Duff cautioned that none are complete models. Most share the drawback of expressing their human tau transgene more prominently in the spinal cord and brain stem than in the cortical, basal forebrain, and other areas that are primarily affected in their corresponding human tauopathy.
One new mouse model is different. Jürgen Götz of the University of Sydney, Australia, presented the first description of a strain that is a surprisingly precise model of early onset human parkinsonism. Lars Ittner in Götz’s group used the cDNA for K369I, a human tau mutation known to cause Pick disease (Neumann et al., 2001. Pick’s is a form of frontotemporal dementia that often produces the movement symptoms of parkinsonism early in the course of disease. For more on Pick’s, see AFTD website). Ittner placed the K369I human tau gene under the control of the mouse Thy1.2 promoter, which drives postnatal expression and thus avoids developmental effects. He obtained mice that overexpress the mutant tau in the substantia nigra in addition to cortex, hippocampus, and amygdala. The substantia nigra is damaged in parkinsonism, but does not express the transgene in most published and widely used tau lines, such as P301L.
In the K369I mice, tau was hyperphosphorylated, and it aggregated with a histopathologic picture of Pick disease in that the lesions are positive for Bielshowsky and negative for Gallyas staining, Götz said. Between 4 and 6 weeks of age, the mice developed all four classic motor symptoms of parkinsonism: a resting tremor, rigidity, bradykinesia (i.e., slowness of voluntary movement), and postural instability. They also responded to treatment with L-dopa, increasing their performance on the beam test. By contrast, the dopamine receptor agonist haloperidol weakened them further. L-dopa worked only in young mice, not in older mice with more advanced disease, said Götz. Together, the symptoms and the drug response mimic parkinsonism in Pick disease, where patients also develop these symptoms early on and benefit from dopaminergic treatment at early but not late stages. Other researchers commented that the drug response might help drug developers validate this mouse model pharmacologically, and make it more practicable for drug screening studies than are some of the existing tau lines.
What causes the early motor symptoms? Aggregation of insoluble tau occurs later, so that can’t be it, Götz said. Already in young transgenic mice, the scientists noted an accumulation of tyrosine hydroxylase (TH), an enzyme needed for dopamine synthesis, in the substantia nigra. Neuronal culture experiments with K369I substantia nigra neurons and human wild-type tau-transfected SH-SY5Y cells indicated that tau impairs the transport of TH such that the enzyme does not reach the neuron’s terminals. In the mice, this happens in the absence of Wallerian degeneration, indicating that mutant tau makes certain neurons dysfunctional long before degeneration sets in. This work adds to a growing body of research on how tau alterations hamper axonal transport (reviewed recently in Lee and Trojanowski, 2006).
Further analysis will show whether, besides parkinsonism, the K369I also have some of the cognitive symptoms of tauopathy. A different new mouse model offers a look at this side of the coin. Luc Buee and colleagues at Inserm and the University of Lille, France, wanted to study the role of tau in Alzheimer disease, where tau pathology occurs in the hippocampus, entorhinal cortex, and other cortical areas. In making a new tau model, these researchers were motivated by the desire to avoid the limb weakness and paralysis that tau mice suffer as a consequence of transgene expression in the spinal cord, because this paralysis can make behavioral testing well nigh impossible. Curiously, by using human tau with two different mutations, they generated a model that has none of the early onset parkinsonism seen by Ittner and Götz, but does exhibit the cognitive symptoms of tauopathy seen in AD. Joint first authors Katharina Schindowski and Alexis Bretteville placed a four-repeat tau construct bearing the G274V and the P301S FTD mutations under control of the same promoter Ittner used. This yielded several lines, of which line 22 is published (Schindowski et al., 2006). These Thy-Tau22 mice express increasing amounts of abnormal tau with age, in entorhinal cortex, CA1 subfield of hippocampus, amygdala, ventral thalamic nuclei, and other cortical areas but not in spinal cord. Abnormal tau first appeared in axonal tracts and later in cell bodies of neurons, and by 12 months the mice showed the full complement of tau pathology typical for AD. Starting at that age, the mice lost neurons in the hippocampus and showed a mild astro- and microgliosis. They had a deficit in basal synaptic transmission but not in LTP. They had no motor deficits but showed cognitive impairments in behavioral tests of anxiety, spatial learning, and spatial memory retention by 6 months of age. In toto, the Thy-Tau22 mice appear to model the tau component of AD, the authors argue.
In Salzburg, Buee presented follow-on data to suggest that these mice have changes in neurotrophic factors that mirror those seen in postmortem AD brain. The mice had a loss of BDNF mRNA and protein in hippocampus and cortex. Buee's group observed an inverse correlation between the AT100-immunoreactivity that indicates tau pathology and BDNF expression. However, keeping the mice in an enriched environment, which is known from other research to induce BDNF, restored BDNF expression even in brain regions strongly affected by neurofibrillary degeneration.
In short, these two new mouse strains hand scientists new tools to explore either the parkinsonian or the cognitive ends of the broad spectrum of clinical symptoms attributed to tau in various neurodegenerative diseases. Colleagues interested in obtaining mice, please contact the scientists directly at jgoetz@med.usyd.edu.au or buee@lille.inserm.fr.—Gabrielle Strobel.
No Available Comments
The mythical Hydra was virtually indestructible. Lop off one of its many heads, and another would take its place. Chopping off the first two N-terminal amino acids of Aβ1-40/42 exposes a glutamic acid residue that can be cyclized to pyroglutamate. That may not sound sinister, but evidence shows that pyroglutamate-derived variants are some of the most stable Aβ peptides and, with increased hydrophobicity, may seed growth of Aβ aggregates. Rather than destroying Aβ, truncating its head might have the opposite effect, unleashing a tenacious pathological cascade. For this reason, truncated Aβ, and pyroglutamate derivatives in particular, have come in for increased scrutiny in recent years, as exemplified by several presentations at last month’s 8th International Conference AD/PD in Salzburg, Austria.
Truncated Aβ as Early Markers of Disease
The existence of N-terminal variants of Aβ has been known for some time. Truncated, or “ragged,” N-termini were described as early as 1993 (see, e.g., Roher et al., 1993). “Now we know that at the N-terminus, Aβ can be truncated in many positions. 2, 3, 4, 5, 7, 8, 9, 10, 11, and almost all of the potential truncated species are found in the brain” said Luc Buee, of INSERM in Lille, France, in an interview. “This is, in part, thanks to work from other groups, including Takaomi Saido’s, who reported pyroglutamate variants, and Virginia Lee and John Trojanowski’s, who reported truncated versions,” he said. Buee and colleagues, including Andre Delacourte, also in Lille, and Kaj Blennow at Goteborg University, Sweden, proposed that these N-terminal truncated species of Aβ may represent good preclinical markers of AD. When they analyzed control brains, they detected various N-truncated forms of Aβ, in addition to a modicum of amyloid deposits and neurofibrillary tangles, in some cases (see Sergeant et al., 2003), suggesting that these species of Aβ are around during the very early stages of pathology. “When there are no neurofibrillary tangles or amyloid deposits, then we don’t see any truncated species, so their presence seems highly specific to preclinical dementia,” Buee said.
These researchers subsequently found N-truncated Aβ in the CSF and plasma of MCI patients. Importantly, their data suggest that truncated Aβ is primarily found in MCI patients who go on to develop full-blown AD (see Vanderstichele et al., 2005). If confirmed by larger studies, truncated Aβ could become the basis of a prognostic test for MCI patients. But Buee and colleagues are also interested in these truncated peptides as potential targets for immunotherapy. As Eugeen Vanmechelen, Innogenetics NV, Gent, Belgium, Buee, and coauthors reported in Salzburg, several truncated Aβ peptides, including N-truncated 4-42 and N-truncated 8-42 Aβ are present at the earliest stages for neuropathology and may represent the earliest pathological forms of the peptide.
Truncated Aβ Fate
Later on in the disease, these truncated species are less abundant than full-length Aβ. By then, the shorter peptides may have already wreaked havoc since they are more hydrophobic than full-length counterparts and may seed the growth of Aβ oligomers and aggregates, researchers believe. “I think truncated forms are very important for nucleation, but once the process is started then aggregation continues with full-length Aβ1-42,” suggested Buee.
Many species of truncated Aβ are quickly degraded. Takaomi Saido’s group at the RIKEN Brain Science Institute, Wako, Japan, has studied the fate of different Aβ derivatives and reported findings in Salzburg. The researchers tracked degradation of various radiolabeled Aβ species after injecting them into rat hippocampus, and found that species truncated at the 2, 3, and 4 N-terminal amino acid were degraded two to three times faster than full-length Aβ. Interestingly, while co-injection of a neprilysin inhibitor, thiorphan, completely blocked degradation of full-length Aβ, it had no such effect on the truncated species. “It appears that truncation makes Aβ susceptible to digestion by some other, as yet unknown, proteases,” suggested Saido in an interview with ARF.
Pyroglutamate-derived species are, however, the exception to this general trend. Saido and colleagues found that N3 pyroglutamate Aβ42 (N3pGlu42) was about three times more stable than Aβ1-42 and almost 10 times more stable than truncated forms without the cyclized glutamate. Given their stability and their hydrophobicity, these pyroglutamate peptides may be particularly dangerous in the brain. “This [N3pGlu42] is actually a major species of Aβ that you find in AD brain,” said Saido (see Piccini et al., 2005). “However, as Steve Younkin’s and other groups have shown, in APP transgenic mice there is almost no pGlu-Aβ42, so the major difference between APP transgenic mice and human AD brains is in the structure of Aβ,” he said (see Kawarabayashi et al., 2001 and Guntert et al., 2006).
Does the lack of N3pGlu42 explain why transgenic mice do not fully recapitulate human AD pathology? To investigate this question, Saido and colleagues generated APP constructs that lack the aspartic acid and alanine residues in positions 1 and 2 of Aβ and ferried them into cortical neurons with viral vectors. Primary cultures of these neurons generate abundant N3pGlu42, indicating that cyclization can occur in the rodent cells (see Shirotani et al., 2002). There are indications that cyclization may exacerbate pathology in whole animals. Saido and colleagues used a slightly different approach to test this possibility. They recently reported that knocking out neprilysin exacerbates Aβ pathology, dampens synaptic plasticity, and compromises cognitive function in APP23 transgenic mice, which produce full-length Aβ42 (see Huang et al., 2006). Saido told ARF that when his group crossed APP transgenic mice with neprilysin knockout mice, they then were able to detect N3pGlu42 in the mouse brain. “This agrees with the notion that when neprilysin activity is lost, N-terminal truncation of Aβ and conversion to pyroglutamate 42 can occur,” said Saido. This idea is in keeping with the potential neprilysin proteolytic site between the third Aβ amino acid, glutamate, and the fourth Aβ amino acid, phenylalanine. Neprilysin should remove the first three amino acids, precluding formation of N3pGlu-Aβ.
The relationship between neprilysin and the formation of pyroglutamate derivatives of Aβ suggests a potential means to control Aβ toxicity. “One way to facilitate Aβ degradation is to upregulate neprilysin, but the other way may be to inhibit cyclization of glutamate,” Saido suggested. The latter is what a biotech company called Probiodrug AG, based in Sachsen-Anhalt, Germany, has in mind.
Prygoglutamate Aβ and Toxicity
Cyclization of glutamate was once thought to be a spontaneous chemical reaction, but that has since been discounted. “In vitro, to convert glutamic acid to pyroglutamine you need to heat the peptide to 135 degrees centigrade in the presence of equimolar water, so it must be an enzymatic process in vivo,” said Saido. In 2004, that is what Probiodrug’s Hans-Ulrich Demuth and colleagues found (see Schilling et al., 2004) .
The enzyme in question is glutaminyl cyclase (QC). QC normally catalyzes the cyclization of N-terminal glutamine, but Demuth and colleagues showed that it can also catalyze, albeit more slowly, the cyclization of N-terminal glutamate. In Salzburg, the scientists reported that, in HEK 293 cells, Aβ3-40 is converted to N3pGlu40 in cells co-transfected with a QC expression construct, and that a QC inhibitor blocked this conversion (see also Cynis et al., 2006). Similarly, the researchers showed that pyroglutamate-derived peptides formed after injecting Aβ3-40 into the cortex of rat brain but were also prevented by the QC inhibitor. And though the amount of N3pGlu40/42 peptides formed in Tg2576 mice is small, Demuth and colleagues demonstrated a dose-dependent reduction in the levels of these peptides upon administration of the cyclase inhibitor (see Bucholz et al., 2006.
Is targeting this cyclase a valid therapeutic strategy? Demuth and colleagues are banking on it. “If you look at the percentages, the plaques in human brain are almost entirely pyroglu-positive—50 to 70 percent if you do a whole plaque analysis. This fact has been ignored,” Demuth told ARF (see also Saido et al., 1995). “Furthermore, if you analyze the core plaques, which are more difficult to dissolve, then you find almost 100 percent of Aβ is pyroglu. So pyroglu forms seem to be the majority of these post-Aβ peptides. It looks like pyroglu peptides are formed early, because they are concentrated in the core and less so in the periphery of the plaques,” he said. This, he says, fits with the faster aggregation kinetics of N3pGluAβ than that of full-length Aβ1-40/42 (see Schilling et al., 2006). In Salzburg, Demuth and colleagues also presented preclinical data suggesting that Tg2576 animals treated with a QC inhibitor for 6 months improved their performance in a contextual fear condition model of learning and memory.
Pyroglutamate Aβ and Pathology
It is not yet clear how pyroglutamate derived Aβ species may be toxic, but they do have a greater propensity to aggregate (see, for example, He and Barrow, 1999; Schilling et al., 2006), which suggests that they might seed formation of larger oligomeric species. There are also indications that N3pGluAβ forms inside neurons and that it might disrupt axonal transport (see Wirths et al., 2006 and Alzforum Webinar by Thomas Bayer, Saarland University, Germany). Intraneuronal Aβ toxicity has become a subject of focused research in recent years (see ARF related news story). Extraneuronal N3pGluAβ is also neurotoxic, as reported by Thierry Pillot and colleagues from Lipidomix, Vandoeuvre-l`es-Nancy, France, in last week’s Neurobiology of Aging online.
If pyroglutamate-derived Aβ peptides turn out to be a major pathological species, then how they form could become central to AD pathology. N3pGluAβ could be derived from Aβ1-40/42 following removal of the first two amino acids. The enzyme dipeptidyl peptidase is a likely candidate for that reaction, suggested Saido, but there may be more to the story. “We believe that the pathway by which pyrogluAβ is formed in the human brain is different from the BACE1-mediated pathway,” said Demuth.
Heterogeneous β-cleavage of APP is not a new idea. Cleavage between the aspartic acid and alanine at positions 1 and 2 of Aβ yields Aβ2-42, for example (see Wiltfang et al., 2001). In Salzburg, Demuth and colleagues showed that HEK293 cells transfected with APP carrying the BACE-1-sensitive Swedish mutation (which substitutes an asparagine/leucine dipeptide for lysine/methionine at the β-secretase site), generated mostly Aβ1-40/42 and barely any N3pGluAβ. Cells transfected with both the Swedish and London mutations showed the same picture. In contrast, cells transfected with wild-type APP, or APP carrying only the London mutation (which substitutes an isoleucine for valine near the γ-secretase site far from the β-secretase site) generated high amounts of N3pGluAβ. The data suggest that there may be differences in APP processing that may be relevant to the treatment of sporadic and different familial forms of AD. In this regard, it is interesting that Pierluigi Gambetti and colleagues at Case Western Reserve University, Cleveland, Ohio, recently reported that N3- and N11-truncated forms of Aβ, which have cyclizable-N-terminal glutamate residues, are more abundant in AD patients carrying PS1 mutations (see Russo et al., 2000).
Demuth and colleagues believe that pyroglu forms of Aβ are generated in the secretory pathway. In Salzburg, they showed data to suggest that both QC and APP co-localize in secretory compartments in the brain. This localization of QC would be in keeping with what may be its primary physiological role, namely to cyclize glutamine residues at the N-terminal of thyrotrophin-releasing hormone (TRH) and other endocrine hormones that mature in secretory vesicles. The slightly acidic pH of the secretory pathway creates the right conditions for QC-catalyzed conversion of glutamate on Aβ, said Demuth. The pH optimum for glutamine cyclization, on the other hand, is slightly more alkaline. Demuth believes that these different pH optima can be exploited to develop a QC inhibitor that blocks formation of pyroglutamate Aβ while having less of an impact on glutamine cyclization. Another reason he is optimistic about this approach is grounded in enzyme kinetics. Because the affinity of QC for glutamine is two orders of magnitude higher than its affinity for glutamate, glutamine cyclization is less likely to be affected by QC inhibition.
On a final note, there may be an endogenous way of ridding the brain of pyroglutamate derivatives of Aβ that might warrant further exploration. In Salzburg, JM Martinez-Marto and colleagues from the University of Jaen, Spain, reported that the enzyme pyrrolidone carboxyl peptidase, which hydrolyzes N-terminal pyroglutamyl residues, can protect neuroblastoma cell lines against the toxicity of pyrogluAβ forms.—Tom Fagan
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.