Flashy Treatment Synchronizes Neurons, Lowers Aβ in Mice
Quick Links
Could staring into a strobe light prevent or treat Alzheimer’s disease? A paper in the December 8 Nature hints as much. Scientists led by Li-Huei Tsai and Edward Boyden from MIT used both optogenetics and a noninvasive light-pulse treatment to restore fading gamma brain waves in an AD mouse model. Doing so quickly quashed soluble Aβ in the brain, slowly dispersed plaques, and even reduced phosphorylated tau levels in a mouse model of tauopathy. These gamma oscillations seemed to reduce the production of Aβ, while at the same time spurring microglia to consume the peptide.
“This is one of the strongest cases made so far for the importance of neuronal network activity in modulating Aβ and tau,” wrote Marc Aurel Busche, Massachusetts General Hospital, Boston, to Alzforum (see full comment below).
Networks of neurons in the brain fire in a coordinated fashion, generating rhythmic waves of electric activity that occur at different frequencies. In the 20-50 Hz range, gamma oscillations are involved in memory encoding and retrieval, perception, and attention. Scientists have reported that gamma oscillations become disrupted in animal models of AD and in people with the disease (May 2012 news; Uhlhaas et al., 2006; Koenig et al., 2005). Conventional wisdom holds that amyloid plaques and neurofibrillary tangles damage the networks that drive these oscillations. However, some reports hint that changes to synaptic activity can, in turn, influence pathology. In AD, overactive synapses spew out Aβ, leading to plaques (see Dec 2005 news; Cirrito et al., 2005; Bero et al., 2011). Tsai and colleagues wondered whether faltering gamma waves might contribute to pathology.
To find out, co-first authors Hannah Iaccarino and Annabelle Singer used electrophysiological recordings to detect gamma oscillations in the hippocampus of three-month-old wild-type and 5XFAD mice. Gamma power in the latter was weaker. Since this was before 5XFAD animals developed amyloid plaques or problems with learning and memory, it suggested that disturbances in gamma waves come prior to plaques.
Could restoring gamma power prevent pathology? The MIT researchers used a virus to express the light-sensitive channelrhodopsin-2 in fast-spiking parvalbumin-positive (FS-PV) interneurons of the CA1 region of the hippocampus in 5XFAD mice. These inhibitory interneurons help generate gamma waves. Through an implanted fiber-optic wire, they pulsed light at 40 Hz to open the rhodopsin channels and drive gamma waves. This does increase overall activity and, at the same time, synchronize it. After an hour, total Aβ40 and Aβ42 in the hippocampus fell by 53 and 45 percent, respectively. Slower or random light pulses did not cause a drop. In fact, random pulses that caused the same average uptick in neuronal activity as the gamma-inducing pulses, but no coordinated gamma oscillations, caused Aβ levels to increase (see image below). That suggests that something about inducing gamma waves, even if overall neural activity goes up, lowers Aβ.
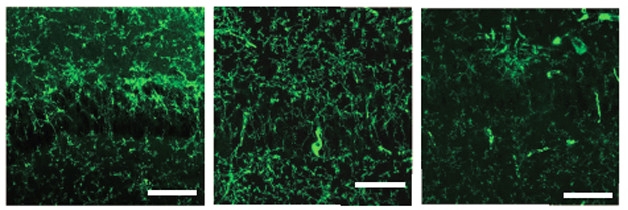
Adios, Amyloid: Soluble Aβ peptides (green) pervade the CA1 region of the 5XFAD mouse brain (left). One hour of stochastic optogenetic stimulation increases the peptide (middle), while eliciting gamma oscillations in this region clears it (right). [Iaccarino et al., Nature.]
Boosting gamma oscillations seemed to both reduce Aβ production from APP and enhance its clearance by microglia. Restoring gamma waves shrank neuronal endosomes in the hippocampus and curtailed intermediates of APP cleavage, including its C- and N-terminal fragments. Microglia doubled in number, swelled up, and became rounded—signs that they were prepping for phagocytosis. The percentage of Aβ-positive microglia almost tripled (see image below). RNA sequencing revealed that microglia upregulated certain genes, many involved in phagocytosis, the authors report.
Next, the researchers wanted to drive gamma oscillations in a less invasive way. They placed three-month-old 5XFAD mice in a dark chamber and, for one hour, flashed a dim LED light at their eyes at 40 Hz to generate gamma frequency waves in the visual cortex. Lo and behold, both Aβ42 and Aβ40 levels dropped in this brain area by 58 percent. This strategy to lower soluble Aβ worked in APP/PS1 and wild-type mice, as well. In six-month old 5XFAD mice that had deposited plaques, a week of this treatment caused 65 percent of them to vanish. Neither random, 20, or 80 Hz light pulses reduced Aβ. In addition, microglial cell bodies swelled, their processes shortened, and they accumulated more Aβ inside. Interestingly, the same light-pulse treatment also reduced phosphorylated tau in the visual cortex of TauP301S mice by 40 percent, and activated microglia in those mice as well.
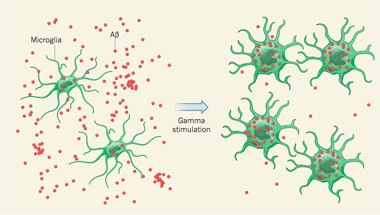
Appetite for Aβ.
After gamma stimulation, microglia devour more of the peptide. [Aron and Yankner, Nature.]
“This is an interesting set of findings. The results suggest new possible ways to lower Aβ levels,” wrote David Holtzman, Washington University in St. Louis, to Alzforum (see full comment below). Current methods enable tracking of Aβ production and clearance in the human central nervous system (CNS), so it could easily be studied if gamma oscillations dial down Aβ levels the way they do in mice, Holtzman wrote. He noted that delta oscillations during slow-wave sleep, as well, are linked with decreased CNS Aβ in both animals and humans (Varga et al., 2016).
The exact mechanism linking synchronized neural activity with microglial activation or APP metabolism is unclear, said Tsai. It appears to involve gene expression changes in microglia that encourage them to phagocytose material. In addition, smaller neuronal endosomes suggest diminished internalization of APP into neurons, which prevents it from being cleaved into Aβ.
Busche proposed an alternative mechanism, whereby enhanced activity of inhibitory neurons may reduce release of Aβ from excitatory neurons. Gamma waves could also improve vascular or glymphatic clearance, he proposed. Busche cautioned that the optogenetically elicited or light-induced gamma rhythms differ from those that occur during normal behavior. He also wondered whether stimulating gamma waves would work later in disease, when interneuron networks have deteriorated (Verret et al., 2012).
How might this all translate to human therapies? Tsai and Boyden have co-founded a small Cambridge, Massachusetts-based start-up company called Cognito Therapeutics that aims to conduct clinical trials of light-flicker therapy in people. Though the visual cortex mostly escapes plaque pathology in AD, it is possible that gamma oscillations generated there could propagate to other more affected regions of the brain, said Tsai. Other technologies, such as deep-brain stimulation and transcranial magnetic stimulation, could be adapted to produce gamma waves, though to her knowledge they haven’t been used yet for that purpose, she said.
“This paper opens a new dimension in the neuroscience of Alzheimer’s disease,” said Bruce Yankner, Harvard Medical School, who penned an accompanying News and Views. “I expect there will be a lot of activity following up on it.”—Gwyneth Dickey Zakaib
References
News Citations
- Needs Salt: Reduced Sodium Channel Linked to Seizures in AD Model
- SfN: Where, How Does Intraneuronal Aβ Pack Its Punch? Part 2
Research Models Citations
Paper Citations
- Uhlhaas PJ, Singer W. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron. 2006 Oct 5;52(1):155-68. PubMed.
- Koenig T, Prichep L, Dierks T, Hubl D, Wahlund LO, John ER, Jelic V. Decreased EEG synchronization in Alzheimer's disease and mild cognitive impairment. Neurobiol Aging. 2005 Feb;26(2):165-71. PubMed.
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005 Dec 22;48(6):913-22. PubMed.
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci. 2011 Jun;14(6):750-6. Epub 2011 May 1 PubMed.
- Varga AW, Wohlleber ME, Giménez S, Romero S, Alonso JF, Ducca EL, Kam K, Lewis C, Tanzi EB, Tweardy S, Kishi A, Parekh A, Fischer E, Gumb T, Alcolea D, Fortea J, Lleó A, Blennow K, Zetterberg H, Mosconi L, Glodzik L, Pirraglia E, Burschtin OE, de Leon MJ, Rapoport DM, Lu SE, Ayappa I, Osorio RS. Reduced Slow-Wave Sleep Is Associated with High Cerebrospinal Fluid Aβ42 Levels in Cognitively Normal Elderly. Sleep. 2016 Nov 1;39(11):2041-2048. PubMed.
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012 Apr 27;149(3):708-21. PubMed.
External Citations
Further Reading
Papers
- Varga AW, Wohlleber ME, Giménez S, Romero S, Alonso JF, Ducca EL, Kam K, Lewis C, Tanzi EB, Tweardy S, Kishi A, Parekh A, Fischer E, Gumb T, Alcolea D, Fortea J, Lleó A, Blennow K, Zetterberg H, Mosconi L, Glodzik L, Pirraglia E, Burschtin OE, de Leon MJ, Rapoport DM, Lu SE, Ayappa I, Osorio RS. Reduced Slow-Wave Sleep Is Associated with High Cerebrospinal Fluid Aβ42 Levels in Cognitively Normal Elderly. Sleep. 2016 Nov 1;39(11):2041-2048. PubMed.
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer's disease: from synapses toward neural networks. Nat Neurosci. 2010 Jul;13(7):812-8. PubMed.
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, Finkbeiner S, Noebels JL, Mucke L. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron. 2007 Sep 6;55(5):697-711. PubMed.
News
- Homing in on Early Alzheimer’s Biomarkers: Does Connectivity Hold the Key?
- Excited Neurons Release More Aberrant Tau
- Shielding Synaptic Glutamate Receptor from Aβ Preserves Memory in Mice
- In mTORC and Theta Rhythms, New Clues to How Sleep Locks Down Memories
- Paper Alert: Microglia Mediate Synaptic Loss in Early Alzheimer’s Disease
Primary Papers
- Iaccarino HF, Singer AC, Martorell AJ, Rudenko A, Gao F, Gillingham TZ, Mathys H, Seo J, Kritskiy O, Abdurrob F, Adaikkan C, Canter RG, Rueda R, Brown EN, Boyden ES, Tsai LH. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature. 2016 Dec 7;540(7632):230-235. PubMed.
- Aron L, Yankner BA. Neurodegenerative disorders: Neural synchronization in Alzheimer's disease. Nature. 2016 Dec 7;540(7632):207-208. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
This study is an ingenious attempt to determine whether interventions that stimulate fast neuronal oscillations in the brain can ameliorate Alzheimer’s pathology. The study contains four key findings:
This is one of the strongest cases made so far for the importance of neuronal network activity in modulating Aβ and tau levels, not only in the diseased brain but perhaps also under physiological conditions (see experiment in wild-type animals). One potential limitation of the study is that the gamma rhythms elicited by optogenetic stimulation or light stimulation may differ from those observed during normal behavior. Nevertheless, the study elegantly extends previous pioneering work demonstrating that neuronal activity is strongly impaired in Alzheimer’s, and that the degree of impairment correlates with the amount of amyloid and tau deposition in the brain (reviewed by Busche and Konnerth, 2016). For example, enhanced action potential firing of excitatory neurons massively increased plaque burden in APP mice (Yamamoto et al., 2015), and reducing neuronal firing rates lowered the amyloid burden (Yuan and Grutzendler, 2016).
The link between increased gamma rhythmicity, microglia activation, and reduced Aβ is intriguing. I wonder what the mechanism is, since microglia are not excitable cells. A plausible hypothesis might be that a gamma-related enhanced level of overall synaptic inhibition, which likely reduces tonic hyperactivity (Busche et al., 2015) and epileptiform activity (Verret et al., 2012) in the brain, results in a reduced release of Aβ from neurons (Cirrito et al., 2005). I wonder whether the evoked gamma rhythm may also improve vascular or glymphatic clearance mechanisms.
This new study implicates a novel treatment approach for AD that is based on the fundamental properties of brain rhythms. Such an alternative approach to conventional drug-based interventions has the advantage that it can remain non-invasive (for example, in the form of biofeedback). However, it is important to keep in mind that increased gamma rhythmicity might transmit information with heightened salience. For example, sensory signals that are normally ignored may instead be misinterpreted by the brain, resulting in misperceptions (e.g., hallucinations). Moreover, the APP mice used in this study were young; thus, a key question is whether in more advanced pathological states, when interneuron networks are more severely impaired (Verret et al., 2012), interventions that stimulate interneurons to generate gamma rhythmicity are still possible.
References:
Busche MA, Konnerth A. Impairments of neural circuit function in Alzheimer's disease. Philos Trans R Soc Lond B Biol Sci. 2016 Aug 5;371(1700) PubMed.
Yamamoto K, Tanei Z, Hashimoto T, Wakabayashi T, Okuno H, Naka Y, Yizhar O, Fenno LE, Fukayama M, Bito H, Cirrito JR, Holtzman DM, Deisseroth K, Iwatsubo T. Chronic optogenetic activation augments aβ pathology in a mouse model of Alzheimer disease. Cell Rep. 2015 May 12;11(6):859-65. Epub 2015 Apr 30 PubMed.
Yuan P, Grutzendler J. Attenuation of β-Amyloid Deposition and Neurotoxicity by Chemogenetic Modulation of Neural Activity. J Neurosci. 2016 Jan 13;36(2):632-41. PubMed.
Busche MA, Grienberger C, Keskin AD, Song B, Neumann U, Staufenbiel M, Förstl H, Konnerth A. Decreased amyloid-β and increased neuronal hyperactivity by immunotherapy in Alzheimer's models. Nat Neurosci. 2015 Dec;18(12):1725-7. Epub 2015 Nov 9 PubMed.
Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012 Apr 27;149(3):708-21. PubMed.
Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005 Dec 22;48(6):913-22. PubMed.
Washington University
In this work, Iaccarino et al. find that specific manipulation of brain gamma activity rapidly over an hour lowers Aβ40 and 42 levels as well as affects microglial gene expression in such a way as to make them more phagocytic. Over time, increasing gamma activity in 5XFAD mice was able to decrease plaque load, possibly through influencing both Aβ production and microglial function.
This an interesting set of findings. It’s been known for some time that different manipulations of synaptic activity, pre- and postsynaptically, can alter Aβ levels acutely up or down, depending on the manipulation. However, influencing specific oscillations in the brain to see if that influences Aβ levels in such a specific way postsynaptically has not been done.
It is interesting that the effect of gamma oscillations may be occurring via influencing neuronal Aβ production as well as via microglial clearance. The results suggest new possible ways to lower Aβ levels.
On a related topic, work on how the sleep-wake cycle influences Aβ suggests that oscillations in the delta range that are prominent during slow-wave sleep are linked with decreased Aβ in the CNS in both animals and humans. In regard to gamma oscillations, as there are good methods to assess Aβ production and clearance in the human CNS/CSF, it will be interesting to determine if increasing gamma oscillations in the human brain also causes a decrease in Aβ levels as shown here in mice.
UCSF & The Gladstone Institutes
UCSF
Gladstone Institute of Neurological Disease, UCSF
In this study, Iaccarino, Singer, and colleagues identify a novel role for hippocampal and cortical slow gamma oscillatory activity in regulating amyloid and tau pathologies in Alzheimer’s disease (AD). After identifying a deficit in slow gamma coincident with sharp-wave ripples (SWRs) in the hippocampus of 5xFAD mice, the authors show that exogenously driving slow gamma activity in the hippocampus and cortex reduces amyloid and tau pathologies. This study makes a strong case for further investigation of slow gamma oscillatory activity in the Alzheimer’s field.
Strikingly, the hippocampal network deficits shown in this study are remarkably similar to those of an ApoE4 mouse model of AD that we published earlier this year. Specifically, we found that human ApoE4 knock-in (ApoE4-KI) mice have reduced SWR abundance and reduced slow gamma power during these SWRs (Gillespie et al., 2016). Despite the extensive pathological differences between 5xFAD mice, which likely model early onset AD, and ApoE4-KI mice, which better represent late-onset AD, this study found that 5xFAD mice have identical deficits in SWR abundance and associated slow gamma power. Further study and comparison of the underlying mechanisms that cause such a similar network impairment will be critical. The corroboration of this phenotype across disparate AD models increases the likelihood that similar hippocampal network abnormalities might exist in AD patients.
It is important to note the distinction between slow gamma coincident with SWRs in the hippocampus and general slow gamma oscillations. The slow gamma induced by optogenetic stimulation in this study lasted much longer and had much higher consistency than physiological slow gamma activity, which fluctuates constantly in the cortex and is modulated even more rapidly in the hippocampus. While the constant stimulation was clearly effective in clearing amyloid and p-tau, it is unlikely that this exogenous slow gamma could have functionally replaced the SWR-associated slow gamma to rescue the deficit they observed. It is also critical to note the difference between hippocampal slow gamma and cortical slow gamma. Each serves unique purposes in coordinating cell assemblies for specific tasks, thus we should be cautious not to pool the CA1 and visual cortex gamma results together. However, regardless of the different types of slow gamma and their brain locations, this study demonstrates clearly that exogenously driving slow gamma in either hippocampus or cortex reduces amyloid and tau pathologies, suggesting a novel approach to target these pathologies in AD.
Additionally, the beneficial effects of stimulating PV cells further supports the hypothesis that GABAergic interneuron dysfunction and/or death are likely involved in the pathogenesis of AD. Previous work by Verret et al. revealed a deficit in cortical gamma activity caused by PV cell dysfunction (Verret et al., 2012). Similarly, our recent study demonstrated that ApoE4 expression in GABAergic interneurons was responsible for the observed SWR-associated slow gamma deficit (Gillespie et al., 2016). It would be interesting to determine the cellular mechanism behind SWR-associated slow gamma deficits in 5xFAD mice and how it might relate to amyloid and tau pathologies and learning and memory impairments.
This intriguing study opens many doors for future research. First, does SWR-associated slow gamma contribute to an amyloid-clearing effect as the induced slow gamma activity does? Does the power of endogenous slow gamma coincident with SWRs correlate with the extent of amyloid or tau pathology or of cognitive deficits in 5xFAD mice? Does exogenously driving slow gamma interfere with normal brain network activities and functions? This is a great start to an exciting line of inquiry.
References:
Gillespie AK, Jones EA, Lin YH, Karlsson MP, Kay K, Yoon SY, Tong LM, Nova P, Carr JS, Frank LM, Huang Y. Apolipoprotein E4 Causes Age-Dependent Disruption of Slow Gamma Oscillations during Hippocampal Sharp-Wave Ripples. Neuron. 2016 May 18;90(4):740-51. Epub 2016 May 5 PubMed.
Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012 Apr 27;149(3):708-21. PubMed.
Roughly half of patients with AD have psychotic symptoms, i.e., delusions and/or hallucinations. Do the authors think that 40 Hz entrainment in these AD with psychosis patients might perhaps reduce their likelihood of symptoms such as hallucinations? Previously, a study found that 40 Hz stimulation partly disrupts gamma-band synchronization in schizophrenia patients who have frequent auditory hallucinations (see Koenig et al., 2012).
Also, use of antipsychotics in AD patients increases the risk of non-cancer mortality by about 60 percent (Koponen et al., 2016). Thus, it would be great if non-invasive gamma stimulation might reduce the need for antipsychotics.
References:
Koenig T, van Swam C, Dierks T, Hubl D. Is gamma band EEG synchronization reduced during auditory driving in schizophrenia patients with auditory verbal hallucinations?. Schizophr Res. 2012 Nov;141(2-3):266-70. Epub 2012 Aug 12 PubMed.
Koponen M, Taipale H, Lavikainen P, Tanskanen A, Tiihonen J, Tolppanen AM, Ahonen R, Hartikainen S. Risk of Mortality Associated with Antipsychotic Monotherapy and Polypharmacy Among Community-Dwelling Persons with Alzheimer's Disease. J Alzheimers Dis. 2017;56(1):107-118. PubMed.
Tsai Lab
Georgia Tech
Picower Institute of MIT
We thank the commentators for taking the time to write about these findings and Alzforum for providing a discussion forum for discoveries in Alzheimer’s disease research. The commentators bring up some excellent points that we were unable to fully discuss in the paper due to length limitations; therefore we elaborate on them here. We would also like to let researchers know that detailed methods for the LED flicker set-up will be available on the Tsai Lab website shortly.
As both Busche and Holtzman point out, our findings build on previous studies that have shown that Aβ peptide levels are elevated following increases in neural activity and reduced following silencing of neural activity (Cirrito et al., 2003; Bero et al., 2011). Therefore, we used the random stimulation condition to control for overall changes in spiking activity caused by stimulation. We compared multi-unit firing rates during interleaved periods of 40 Hz and random stimulation and found no significant differences between firing rates in these conditions (Ext. Data Fig. 1o). Thus, while the random condition did not induce gamma oscillations, it did result in similar amounts of multi-unit spiking activity (Fig. 1e). Accordingly, we think reduced Aβ levels in response to 40 Hz stimulation were not due to decreased spiking activity.
Because our recordings and analysis did not distinguish between pyramidal cells and FS-PV-interneurons, we cannot exclude that pyramidal cell firing rates differed between these conditions but firing of FS-PV-interneurons or other cell types masked this change. However, random optogenetic stimulation of FS-PV-interneurons provided the same amount of direct stimulation of FS-PV-interneurons yet did not reduce amyloid. In fact, optogenetic stochastic stimulation more than tripled amyloid levels while stochastic visual flicker produced no significant change, which may indicate that some aspects of the random stimulation have neurotoxic effects. While random stimulation did not result in increased gamma power, we noticed a trend of small increases in power in a wide range of frequencies, from around 20 Hz to greater than 60 Hz (Fig. 1 e). These results point to a need to understand how patterns of spiking activity affect molecular pathways and disease pathology.
As Jones, Gillespie, and Huang describe, the deficits we find in the 5XFAD mouse model are very similar to those they found in the ApoE4 mouse and converge with evidence of gamma deficits Verret et al. report in the hAPP mouse model (Verret et al., 2012; Gillespie et al., 2016). This converging evidence from multiple mouse models of AD, including transgenic and knock-in models, suggests that these results are not due solely to overexpression of transgenes or to other side effects particular to one model. Importantly, studies in humans with AD have found altered gamma (Stam et al., 2002). Together, these results from mice and humans suggest that multiple molecular pathways that contribute to Aβ pathology may alter gamma activity in AD.
Further study is needed to determine whether driving gamma oscillations to reduce Aβ will be therapeutic in humans, as Holtzman points out. Broadly, a current prevailing theory of AD pathogenesis points to microglia malfunction, specifically microglia’s failure to clear out pathological molecules, as a key mechanism of disease progression (Heneka et al., 2014). Therefore, interventions that recruit microglia back to an endocytotic state, as 40 Hz stimulation does, have strong therapeutic potential.
In addition, as Busche mentions, the younger mice used in this study likely have intact interneuron networks able to entrain to 40 Hz. It will be important to determine whether methods that drive gamma oscillations will be efficient in patients and mouse models with more severe interneuron network deficiencies. Alternately, a gamma-based therapy may need to be implemented preventatively or prior to break down of interneuron networks. We look forward to a deeper investigation of both the potential and boundaries of this network-based intervention.
References:
Cirrito JR, May PC, O'Dell MA, Taylor JW, Parsadanian M, Cramer JW, Audia JE, Nissen JS, Bales KR, Paul SM, Demattos RB, Holtzman DM. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-beta metabolism and half-life. J Neurosci. 2003 Oct 1;23(26):8844-53. PubMed.
Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci. 2011 Jun;14(6):750-6. Epub 2011 May 1 PubMed.
Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, Mucke L, Palop JJ. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell. 2012 Apr 27;149(3):708-21. PubMed.
Gillespie AK, Jones EA, Lin YH, Karlsson MP, Kay K, Yoon SY, Tong LM, Nova P, Carr JS, Frank LM, Huang Y. Apolipoprotein E4 Causes Age-Dependent Disruption of Slow Gamma Oscillations during Hippocampal Sharp-Wave Ripples. Neuron. 2016 May 18;90(4):740-51. Epub 2016 May 5 PubMed.
Stam CJ, van Cappellen van Walsum AM, Pijnenburg YA, Berendse HW, de Munck JC, Scheltens P, van Dijk BW. Generalized synchronization of MEG recordings in Alzheimer's Disease: evidence for involvement of the gamma band. J Clin Neurophysiol. 2002 Dec;19(6):562-74. PubMed.
Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat Rev Immunol. 2014 Jul;14(7):463-77. PubMed.
Make a Comment
To make a comment you must login or register.