Novel Methylation Distinguishes Neuron Types, May Dictate Disease
Quick Links
Beyond the four-letter alphabet of the genome, a far richer code dictates when and where genes are transcribed. The epigenome—defined by an ever-expanding list of modifications to DNA and the proteins that interact with it—determines which genes are dialed up or down and gives each cell type its unique personality. Thickening an already dense plot, three recent papers suggest that the brain may have its own epigenetic lingo.
One, published in Neuron on June 17, described the epigenome of three different types of neuron from the mouse brain—one excitatory, and two inhibitory. Among a slew of other findings, the study reported that neurons harbor a striking degree of cytosine methylation beyond the well-known cytosine-guanine (CpG) sites. This novel modification more closely correlated with gene expression and with neuronal phenotype than did the more common CpG methylation.
Two other studies—one published in the Proceedings of the National Academy of Sciences on April 28 and the other in Nature on June 4—highlighted the importance of non-CpG methylation in Rett syndrome, a neurodevelopmental disorder accompanied by widespread alterations in gene expression. Researchers found that the protein MeCP2, which causes Rett syndrome when mutated, regulates a plethora of genes by binding to non-CpG methylated DNA that starts to surface in young mice. The findings expand the complex epigenetic code in the brain, and point to master mechanisms that may shape the fate and function of neurons through aging and disease.
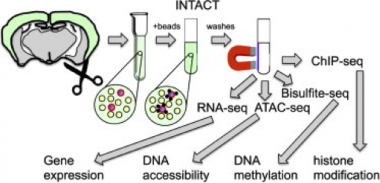
Epic Epigenomics.
Researchers use sorted nuclei from the INTACT method to interrogate all manner of epigenetic marks, ranging from DNA methylation to histone modification. [Image courtesy of Henikoff, Neuron 2015].
In contrast to methylated CpG (mCG), non-CpG methylation, or “mCH,” where H can be an A, T, or C, rarely happens in somatic cells, although it accounts for around a quarter of the methylation in pluripotent stem cells (see Lister et al., 2009 and Ziller et al., 2011). A recent study led by Joseph Ecker at the Salk Institute in La Jolla sequenced the “methylome” of most human organs except the brain. It reported that, though sparse, mCH does occur in adult tissues. There it correlates with gene repression, but likely only in distinct subsets of cells within each organ, (see Schultz et al., 2015). The story is different in the brain, where researchers have found non-CpG methylation to be far more prominent (see Xie et al., 2012, Varley et al., 2013, and Guo et al., 2014).
To generate a more detailed epigenetic map of neurons in the mouse brain, Ecker joined forces with researchers from Jeremy Nathans’s lab at Johns Hopkins University in Baltimore and Sean Eddy’s lab at the Howard Hughes Medical Institute in Ashburn, Virginia. The researchers employed a technique called INTACT (isolation of nuclei tagged in specific cell types) to study nuclei from three types of neuron (see image above). The technique, which uses antibodies to capture nuclei expressing a protein tag, had been established in a plant model, and later used in flies, worms, and frogs, but never in mammals (see Deal and Henikoff, 2010). INTACT isolates nuclei from homogenized tissue that is first frozen intact. This eliminates the need to first separate or sort the different types of cells, which can damage and/or activate neurons and confound results. INTACT allows researchers to obtain enough genetic material from specific cell types to run methylation and other epigenetic analyses.
As outlined in the Neuron paper, the researchers generated three lines of transgenic animal, each expressing a green fluorescent protein (GFP) and Myc-tagged version of the nuclear membrane protein SUN1, in one of three types of neocortical neuron: excitatory pyramidal neurons, parvalbumin (PV)-expressing fast spiking interneurons, or vasoactive intestinal peptide (VIP)-expressing interneurons. PV and VIP interneurons are both inhibitory. After freezing and homogenizing the tissue but leaving nuclei intact, the researchers used anti-GFP and/or anti-Myc antibodies conjugated to magnetic beads to isolate the nuclei from each neuronal type. Nearly 100 percent of the resulting nuclei were derived from the desired cell type, the authors reported.
The researchers then measured the transcriptome and epigenome of the three types of neuron. RNA-Seq analysis revealed more than 4,000 genes expressed at different levels among the three types. Methyl-C sequencing identified a striking amount of non-CpG methylation, which comprised at least 40 percent of total methylcytosines in each neuronal type. Interestingly, 8,000 genes displayed more than a 50 percent difference in mCH levels among the neuronal types.
The mCH distribution more strongly distinguished neuron types than did overall gene expression or mCG patterns. For example, the gene for the VIP-specific transcription factor Prox1 had 23-fold higher mCH in excitatory neurons and 32-fold higher mCH in PV neurons than in VIP neurons. The researchers found that mCH inversely correlated with gene expression. In fact, CH methylation within a gene correlated more highly with repression than inaccessibility of DNA to transcription machinery (as measured by the ability of a transposase to grab onto genes).
In some cases, INTACT clarified discrepancies between gene expression and methylation patterns that had clouded prior experiments analyzing all neuron types at once. For example, although DNA methylation is known to correlate with lower gene expression, the PV-specific Lhx6 gene appeared to be both highly methylated and actively transcribed in samples of whole cortex or isolated bulk neurons. However, when the researchers looked individually at PV versus VIP and excitatory neuronal populations, they found that active transcription and high methylation never occurred in the same cell types: Lhx6 was highly methylated in VIP and excitatory neurons, but not in PV neurons, the only one of the 3 neuron types where it was highly expressed.
The researchers extended their findings beyond the realm of CpG versus CH methylation by linking methylation patterns with other epigenetic marks. They found that regions where DNA is accessible, which are thought to represent active promoter and enhancer regions, varied strongly between neurons. These regions tended to be hypomethylated and have histone modifications known to activate enhancers. When the researchers used methylation patterns or DNA accessibility to put neurons into hierarchical clusters, they found that VIP and PV inhibitory neurons grouped together, and were distinct from excitatory neurons.
Commentators were roundly impressed with the study and enthusiastic about using INTACT in their own research. While the transgenic method cannot be used to study epigenetics in people, Philip de Jager of Brigham and Women’s Hospital in Boston said Nathans’s work established the extent to which epigenomes of neurons vary, and will pave the way for technological advances in human studies. De Jager heads an effort to map the epigenome from post-mortem AD brain samples (see Bennett et al., 2014). Mapping typically involves isolating specific cells by laser capture microdissection, a laborious process that must be repeated thousands of times per sample because a single cell does not yield enough material for methylation and other epigenetic assays. De Jager said that researchers are devising ways to eke out epigenetic profiles from single cells, which would ultimately simplify the comparison of different neuronal types.
Non-CpG Methylation Calls the Shots in Rett Syndrome
Non-CpG methylation may be the new suspect in Rett syndrome, according to the PNAS and Nature papers. As its name suggests, methyl CpG binding protein 2 (MeCP2), the principle cause of this disease, has a penchant for binding methyl-CpG sites, however it has been reported to bind to mCH as well. First linked to Rett syndrome in 1999 by Huda Zogbhi of Baylor College of Medicine in Houston, MeCP2 was deemed a transcriptional regulator that could both activate or repress genes (see Amir et al., 1999 and Chahrour et al., 2008), but researchers have since struggled to understand how the protein affects transcription and causes disease (see Pohodich and Zogbhi, 2015 and Lyst and Bird, 2015). MeCP2 expression begins after birth, and continues to rise throughout postnatal development. Researchers have proposed that this late expression could explain why children with Rett syndrome appear normal at first. Interestingly, a study from Ecker’s lab indicated that non-CpG methylation also rises postnally; however the functional significance of MeCP2’s interaction with mCH had not been explored (see Lister et al., 2013).
Zogbhi led the study reported in PNAS. To make some sense out of MeCP2’s widespread DNA binding pattern, first author Lin Chen and colleagues expressed a fully functional, GFP-tagged MeCP2 in mice lacking any other copy of the gene. They then closely monitored the protein’s binding across the genome using chromatin immunoprecipitation. In addition to binding mCG, MeCP2 also associated strongly with genes containing the highest levels of mCH.
Did MeCP2 binding to mCH regulate gene expression? To find out, the researchers compared transcriptome data from adult wild type mice, MeCP2 knockout mice, or mice overexpressing MeCP2. Transcriptional regulation in the hypothalamus, a region thought to be involved in Rett symptoms, was like two sides of a coin. Genes that were turned up in the knockouts were turned down in the mice overexpressing MeCP2, and vice versa. Surprisingly, whether or not a gene’s expression was regulated by MeCP2 did not depend on its mCG content; rather, genes with high levels of mCH tended to be most affected by alterations in MeCP2 expression.
MeCP2 does not always downregulate genes, however. Researchers previously reported that expression of brain derived neurotrophic factor (BDNF) wanes in MeCP2 knockout mice, and that overexpressing the trophin relieves Rett-like symptoms in these animals (see Chang et al., 2006). What makes BDNF different? Chen and colleagues found that six-week old mice harbored more mCH at the BDNF gene than three-week old mice, and that MeCP2 bound to these mCH sequences. The researchers hypothesized that MeCP2 then upregulates the BDNF gene during development, but mutated MeCP2 does not. This could explain some symptoms of Rett syndrome. They did not test if mutated versions of MeCP2 bound BDNF mCH sequences.
Researchers led by Michael Greenberg of Harvard Medical School tackled the mystery of MeCP2 gene regulation from a different angle. As described in the Nature paper, first author Harrison Gabel started by looking for commonalities between genes that were turned up or down in MeCP2 knockout mice. Genes downregulated in MeCP2 knockout mice, including BDNF, seemed to share no trait. However, Gabel found that a much larger subset of genes was upregulated in the knockouts and that these were five times longer, on average, than a typical gene. Long genes tend to be more actively transcribed in brain than short genes. The same pattern surfaced when the researchers referenced MeCP2-regulated gene lists produced by several other labs. Gabel found that in MeCP2 knockout mice the expression of long genes increased as the mice aged from four to nine weeks old. Mouse lines expressing MeCP2 mutants that cause Rett syndrome also upregulated long genes, and this correlated with the severity of the symptoms. Many of these long genes encoded proteins involved in neuronal functions, such as axon guidance, calcium signaling, and synapse formation.
Why would MeCP2 preferentially regulate long genes? The researchers discovered that, for unknown reasons, those genes have more mCH per nucleotide than do shorter genes. To explore the idea that non-CpG methylation played a part on MeCP2’s regulation of long genes, the researchers generated mice that lack expression of the methyltransferase Dnmt3a in the brain. Dnmt3a methylates non-CpG cytosines but is not required for CpG methylation. In Dnmt3a knockouts, methyl-CA residues were absent while CpG methylation appeared normal. Lo and behold, long gene expression was upregulated similarly to that in MeCP2 knockout mice, suggesting that MeCP2 wields its regulatory influence though binding to mCA residues.
How could the methyl DNA binding protein regulate gene expression? Gabel speculated that MeCP2 molecules act like speed bumps for RNA polymerase. In long genes where mCH residues abound and MeCP2 liberally decorates the DNA, RNA polymerase would have an extraordinarily rough ride and transcription would be inefficient. This fits with MeCP2 modulating transcription incrementally rather than switching it on or off. MeCP2 may also temper gene expression in collaboration with transcriptional repressors, as has been demonstrated by MeCP2’s association with the NCoR/SMRT repressor complex. Some MeCP2 mutations that cause Rett syndrome abolish the interaction with this repressor (see Lyst et al., 2013).
Reconciling the speed bump hypothesis with Zogbhi’s findings that MeCP2 activates some genes, including BDNF, is trickier. It is possible that MeCP2 associates with activators of transcription as well, Gabel said. Furthermore, increased expression of some genes could be an indirect result of suppression of others, said Gabel. Zogbhi said her lab is trying to figure out how MeCP2 affects transcription.
What about the role of mCH in neurodegenerative disease? The jury is still out, but studies are underway. De Jager told Alzforum that preliminary data from comparisons between a small number of post-mortem AD brain samples and normal aging brains revealed no striking differences in mCH. However, this study, which used a microarray to measure methylation at specific sites in the genome, was geared toward CpG methylation and may have missed differences in mCH. De Jager said that focused studies on this particular type of methylation will be needed to determine how the modification changes with age or contributes to disease.
Philip Landfield of the University of Kentucky in Lexington called Nathans’s study “amazing.” He added that patterns of neuronal activity, which fluctuate with age and during disease, are likely the driving force behind epigenetic changes in the brain. In turn, these epigenetic marks may profoundly affect neuronal activity and survival. Disentangling this complex web of regulation is the daunting challenge epigenetic researchers now face.—Jessica Shugart
References
Paper Citations
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, Edsall L, Antosiewicz-Bourget J, Stewart R, Ruotti V, Millar AH, Thomson JA, Ren B, Ecker JR. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009 Nov 19;462(7271):315-22. PubMed.
- Ziller MJ, Müller F, Liao J, Zhang Y, Gu H, Bock C, Boyle P, Epstein CB, Bernstein BE, Lengauer T, Gnirke A, Meissner A. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011 Dec;7(12):e1002389. Epub 2011 Dec 8 PubMed.
- Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, Lin S, Lin Y, Jung I, Schmitt AD, Selvaraj S, Ren B, Sejnowski TJ, Wang W, Ecker JR. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015 Jun 1; PubMed.
- Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, Ren B. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell. 2012 Feb 17;148(4):816-31. PubMed.
- Varley KE, Gertz J, Bowling KM, Parker SL, Reddy TE, Pauli-Behn F, Cross MK, Williams BA, Stamatoyannopoulos JA, Crawford GE, Absher DM, Wold BJ, Myers RM. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013 Mar;23(3):555-67. Epub 2013 Jan 16 PubMed.
- Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, Zhu H, Chang Q, Gao Y, Ming GL, Song H. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014 Feb;17(2):215-22. Epub 2013 Dec 22 PubMed.
- Deal RB, Henikoff S. A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev Cell. 2010 Jun 15;18(6):1030-40. PubMed.
- Bennett DA, Yu L, Yang J, Srivastava GP, Aubin C, De Jager PL. Epigenomics of Alzheimer's disease. Transl Res. 2014 May 16; PubMed.
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999 Oct;23(2):185-8. PubMed.
- Chahrour M, Jung SY, Shaw C, Zhou X, Wong ST, Qin J, Zoghbi HY. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008 May 30;320(5880):1224-9. PubMed.
- Lyst MJ, Bird A. Rett syndrome: a complex disorder with simple roots. Nat Rev Genet. 2015 May;16(5):261-75. Epub 2015 Mar 3 PubMed.
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, Yu M, Tonti-Filippini J, Heyn H, Hu S, Wu JC, Rao A, Esteller M, He C, Haghighi FG, Sejnowski TJ, Behrens MM, Ecker JR. Global epigenomic reconfiguration during mammalian brain development. Science. 2013 Aug 9;341(6146):1237905. Epub 2013 Jul 4 PubMed.
- Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006 Feb 2;49(3):341-8. PubMed.
- Lyst MJ, Ekiert R, Ebert DH, Merusi C, Nowak J, Selfridge J, Guy J, Kastan NR, Robinson ND, de Lima Alves F, Rappsilber J, Greenberg ME, Bird A. Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat Neurosci. 2013 Jul;16(7):898-902. Epub 2013 Jun 16 PubMed.
Further Reading
Papers
- Henikoff S. Epigenomic Landscapes Reflect Neuronal Diversity. Neuron. 2015 Jun 17;86(6):1319-21. PubMed.
- He Y, Ecker JR. Non-CG Methylation in the Human Genome. Annu Rev Genomics Hum Genet. 2015 Jun 4; PubMed.
- Luo C, Ecker JR. Epigenetics. Exceptional epigenetics in the brain. Science. 2015 Jun 5;348(6239):1094-5. PubMed.
Primary Papers
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, Eddy SR, Ecker JR, Nathans J. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron. 2015 Jun 17;86(6):1369-84. PubMed.
- Chen L, Chen K, Lavery LA, Baker SA, Shaw CA, Li W, Zoghbi HY. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc Natl Acad Sci U S A. 2015 Apr 28;112(17):5509-14. Epub 2015 Apr 13 PubMed.
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, Greenberg ME. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015 Jun 4;522(7554):89-93. Epub 2015 Mar 11 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.