Aβ Immunotherapy—Muscling in on Inclusion Body Myositis
Quick Links
Amyloid-β (Aβ) oligomers are presumed guilty of killing neurons in Alzheimer disease, but that may not be their only transgression. In the muscle-wasting disease inclusion body myositis (IBM), intracellular Aβ-containing deposits pepper the scene of the crime, where the victims are not neurons, but muscle fibers. The parallels to AD are clear: affected muscle tissue is subject to inflammation and deposits of other proteins such as tau, ApoE, and α-synuclein. Three years ago, Masashi Kitazawa in Frank LaFerla’s lab at the University of California at Irvine published a mouse model of IBM, in which APP and presenilin transgenes drive Aβ42 accumulation in muscle cells (Kitazawa et al., 2006). Now, the same researchers report that Aβ immunization can reduce intracellular Aβ deposits and motor dysfunction in the IBM mice. The work, published this week in the Journal of Neuroscience, supports the hypothesis that Aβ oligomers are the toxic force in IBM. The work also holds out hope that Aβ immunization, now in clinical testing for AD, may offer a novel therapeutic option for this currently untreatable disease.
IBM is rare, affecting just 1 to 10 per million people, but it is still the most common cause of muscle degeneration in people over 65. Work over the last decade, most prominently by Valerie Askanas at the University of Southern California in Los Angeles has implicated Aβ accumulation, and in particular Aβ42, as a cause of the muscle loss (Vattemi et al., 2009).
To make a mouse model of IBM, Kitazawa used the muscle creatine kinase promoter to target overexpression of the amyloid precursor protein (Swedish mutation) to muscle cells. Addition of the presenilin1 M146V mutant caused overproduction specifically of Aβ42, and the animals developed key features of IBM including intracellular Aβ accumulation in muscle fibers, inflammation, tau deposits, muscle degeneration, and motor deficits.
Because the mice showed motor impairment proportional to the level of muscle Aβ42, the investigators wanted to know if removing Aβ42 would affect progression of disease. They immunized the mice with a multivalent Aβ1-33 peptide conjugate, which induced a strong antibody response. After three months of immunization, the mice showed improved motor skills, as indicated by their ability to balance on a rotating rod. In muscle tissue, there was less Aβ, the cells appeared less vacuolated, and they expressed less αB-crystallin, a marker of cell stress. There was no change in the inflammatory marker MHC type I. Staining of cells indicated that antibodies could enter muscle fibers and bind Aβ deposits there (see image), leading to their clearance by unknown mechanisms.
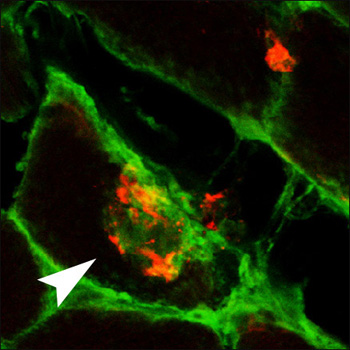
Muscling in on Aβ
An Aβ deposit (red) in muscle fibers from a mouse model of inclusion body myositis also stains with anti-mouse IgG antibodies (green) after immunization of the animals with Aβ. View larger image. Image credit: Frank LaFerla and the Journal of Neuroscience
The positive effects of immunization seemed to be due to the removal of Aβ oligomers. Blots of muscle tissue with either of two oligomer-specific antibodies, A11 or OC, showed reduction of oligomers in immunized mice. Finally, Kitazawa showed that addition of oligomers to muscle cells in culture was directly toxic, and that the adverse effects could be prevented by adding antibodies from immunized mice. From this, the investigators conclude that immunization may act by neutralizing oligomers’ toxicity.
“In this study we provide evidence that Aβ is one of the key pathological components in IBM and promoting clearance of Aβ may be a valid therapeutic strategy to pursue,” the authors write. “We’re hoping this would be a basis for doing a clinical trial with immunotherapy,” LaFerla told ARF. That will depend on the ongoing studies of vaccines for AD, to prove that the vaccines are safe. In addition, LaFerla pointed out that because IBM is so rare, it is hard to convince pharma that the studies are worth doing. (For more on the potential of Aβ vaccines in IBM, see comment below by Askanas).
Outstanding questions include the mechanism of clearance of Aβ. One difference between AD and IBM is the location of the Aβ deposits. In AD, amyloid is mostly extracellular, while in IBM it is mostly intracellular. However, there are indications that intracellular Aβ may also be important in AD, and recent studies have shown that antibodies are capable of clearing intracellular proteins, including Aβ (Tampellini et al., 2007) and α-synuclein (see ARF related news story and Masliah et al., 2005). In the case of the IBM mice, the researchers have no idea yet what happens to the Aβ after antibody binding, LaFerla said.
“This work says that Aβ oligomers are toxic to a wide variety of cell types, both muscles and neurons,” LaFerla told ARF. How might oligomers kill muscle cells? “Our hypothesis is that Aβ is acting in a similar fashion to what it does in neurons. If we had to guess, we’d say it probably interferes with protein disposal systems, autophagic processes, and the proteasome,” LaFerla said. That is a subject of active investigation, he added.—Pat McCaffrey
References
News Citations
Paper Citations
- Kitazawa M, Green KN, Caccamo A, Laferla FM. Genetically augmenting Abeta42 levels in skeletal muscle exacerbates inclusion body myositis-like pathology and motor deficits in transgenic mice. Am J Pathol. 2006 Jun;168(6):1986-97. PubMed.
- Vattemi G, Nogalska A, King Engel W, D'Agostino C, Checler F, Askanas V. Amyloid-beta42 is preferentially accumulated in muscle fibers of patients with sporadic inclusion-body myositis. Acta Neuropathol. 2009 May;117(5):569-74. PubMed.
- Tampellini D, Magrané J, Takahashi RH, Li F, Lin MT, Almeida CG, Gouras GK. Internalized antibodies to the Abeta domain of APP reduce neuronal Abeta and protect against synaptic alterations. J Biol Chem. 2007 Jun 29;282(26):18895-906. PubMed.
- Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005 Jun 16;46(6):857-68. PubMed.
Other Citations
{kind=link}
Further Reading
Primary Papers
- Kitazawa M, Vasilevko V, Cribbs DH, Laferla FM. Immunization with amyloid-beta attenuates inclusion body myositis-like myopathology and motor impairment in a transgenic mouse model. J Neurosci. 2009 May 13;29(19):6132-41. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Southern California
Sporadic inclusion-body myositis (s-IBM) is the most common progressive muscle disease of older individuals (Askanas and Engel, 2008). Accumulation of amyloid-β protein, and especially of its longer fragment Aβ42, appears to play a crucial role in s-IBM pathogenesis. In this paper Kitazawa et al. report interesting findings, using one of their transgenic mouse models overexpressing human Aβ precursor protein (AβPP). This model resembles some pathologic aspects of s-IBM, including the presence of Aβ42 and phosphorylated-tau protein.
In this transgenic mouse model, immunization with Aβ was reported to attenuate the motor impairment and also to reduce the load of Aβ and Aβ oligomers in muscle. Accordingly, the authors proposed that “there may be real opportunities to safely pursue a clinical trial in IBM patients.”
However, the reportedly improved mice are not human, and cautions are necessary in attempting to translate those results into therapeutic trials in s-IBM patients. A previous study involving Aβ immunization of Alzheimer disease patients was not beneficial, and was stopped because of dangerous meningoencephalitis side effects. Several other human immunization trials in Alzheimer disease are currently in progress, but their safety and efficacy are not yet known (to my knowledge).
s-IBM is a progressive muscle disease gradually leading to severe disability from muscle weakness, but it does not appear to affect the patients’ cognitive function or lifespan. s-IBM patients can enjoy a long, productive life, intellectually and socially. Before planning a trial of Aβ immunization in s-IBM patients, the ongoing trials in Alzheimer disease must have demonstrated long-term safety. To develop a therapeutic Aβ immunologic approach to s-IBM patients would be quite a challenge. One would need to achieve an enduring therapeutic clearance of toxic Aβ oligomers located within muscle fibers without inducing widespread cytodestructive autoimmune reactions.
References:
Askanas V, Engel WK. Inclusion-body myositis: muscle-fiber molecular pathology and possible pathogenic significance of its similarity to Alzheimer's and Parkinson's disease brains. Acta Neuropathol. 2008 Dec;116(6):583-95. PubMed.
University of Kentucky, Sanders-Brown COA
Dr. Askanas raises some excellent points. I would add that there is still substantial resistance within the field to the idea that Aβ42 drives s-IBM, with a large number of researchers holding that the disease is purely an inflammatory myopathy. This has always struck me as puzzling, given that treatments targeting the immune system have been widely ineffective. Moving forward with a clinical trial will require overcoming this resistance. This situation shares an interesting parallel with AD, in that the success (or failure) of agents targeting Aβ42 will go a long way towards determining the true role of amyloid in the disease.
Make a Comment
To make a comment you must login or register.