Lipoprotein Receptor Deficiency and Aβ—Spiraling Toward Alzheimer's?
Quick Links
Researchers have connected the dots in a pivotal pathway that they believe may tip the balance toward Alzheimer’s disease. In a study published September 17 in Neuron, Guojun Bu at the Mayo Clinic in Jacksonville, Florida, and colleagues report that a drop in the expression of the low-density lipoprotein receptor-related protein 6 (LRP6) damages neuronal synapses, weakens memory in mice, and ramps up the production of Aβ. In turn, Aβ dials down LRP6, promoting a vicious cycle. The findings may bolster support for LRP6 and members of downstream signaling pathways as tractable therapeutic targets for AD.
“The idea of the vicious pathogenic cycle in AD has been out there forever,” said Robert Vassar of Northwestern University in Chicago, who was not involved in the study. “But I think this is the first clear and convincing example of a molecular mechanism that really starts to get at how Aβ stimulates its own production.”
LRP6 is a receptor for Wnt signaling, which plays a variety of important roles in the development and function of the nervous system. Working in concert with its co-receptor, Frizzled, LRP6 responds to activation by one of several Wnt ligands to initiate a signaling cascade that stabilizes cytoplasmic β-catenin. This factor then moves into the nucleus and targets a slew of genes (see image below). Wnt signaling reportedly promotes synaptic function and plasticity in adult animals (see Cerpa et al., 2008; Cerpa et al., 2010; Inestrosa et al., 2014; and Purro et al., 2014).
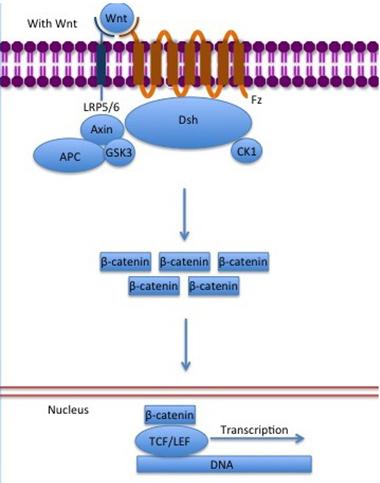
Pathway to Protection.
LRP6 teams up with Frizzled (Fz) to deliver Wnt signals, which liberate β-catenin and lead to changes in gene regulation. The pathway may play a key role in protecting neurons from Aβ-mediated destruction. [Modified image courtesy of Gpruett2, Wikimedia Commons.]
Genetic association studies have linked LRP6 to AD, including a splice variant that tones down Wnt signaling (see May 2007 news story; Alarcon et al., 2013). In the present paper, first author Chia-Chen Liu and colleagues hypothesized that a loss of LRP6 function could be an early player in promoting neurodegeneration.
“Because we know that synaptic dysfunction and amyloid buildup happen before the clinical onset of Alzheimer's, we are very interested to identify the molecular mechanism that may contribute to this pathogenic process,” Liu told Alzforum.
Liu and colleagues started by generating a conditional knockout mouse (LRP6 cKO) that lacks LRP6 expression only in forebrain neurons. The mice displayed deficits in associative memory by 22 months of age. They froze in place less often when returned to an environment where they had received a foot shock, or when cued with a sound that had accompanied that zap.
To pursue the cause of the cognitive decline, the researchers next measured long-term potentiation (LTP), a yardstick of synaptic plasticity, in neurons from hippocampal slices. While hippocampal neurons from control mice stayed primed for action long after stimulation, those from LRP6 cKO mice relaxed a lot sooner. They also had fewer dendritic spines on cortical and hippocampal neurons than normal mice. Synapses in those cells contained less PSD-95, a postsynaptic scaffold protein, and less GluR1, a key subunit of glutamate neurotransmitter receptors. Neuroinflammation plagued these animals as well. Inflammatory cytokines, activated astrocytes, and activated microglia all ticked up in the hippocampus. These phenotypes emerged after 18 months of age, suggesting that LRP6 deficiency triggers age-related neuronal damage.
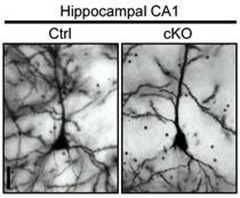
Communication Breakdown.
Mice lacking LRP6 expression in the forebrain had fewer dendritic spines in their hippocampus (right) than normal animals (left). [Image courtesy of Liu et al., Neuron 2014.]
To check whether LRP6 interacts with amyloid pathology, the researchers crossed the LRP6 conditional knockouts with APP/PS1 mice, which develop amyloid plaques. By six months of age, the crosses had amassed more plaques than APP/PS1 controls, and this difference grew by nine months. The researchers concluded that in addition to directly affecting neuronal function, LRP6 helps prevent amyloidosis. By 12 months, the crosses became forgetful as measured by the same foot-shock recall test.
How might LRP6 increase plaque load? Immunoprecipitation experiments suggested that LRP6 and APP bind each other via their extracellular domains. Does this interaction alter APP processing? The researchers quantified different APP products in the mouse neuroblastoma cell line N2a, with APP overexpressed and LRP6 knocked down. They found elevated levels of the β-secretase products APPβ and CTFβ, indicating more amyloidogenic processing of APP when there is an excess APP over LRP6.
More APP dotted the surface in cells that expressed LRP6 than in those that did not, suggesting that LRP6 somehow reroutes APP to this subcellular location. Interestingly, previous studies had reported that β-secretase activity predominates in endosomes over the cell surface, where α-secretase abounds. By shuttling APP to the cell surface, LRP6 could downregulate amyloidogenic processing, suggested the authors. Levels of β-secretase products also rose in the APP/PS1-LRP6 knockouts.
LRP6 affected processing of APP, but how about vice versa? Could APP’s products affect LRP6? The researchers addressed this by dosing neuroblastoma cells with Aβ42 oligomers prepared in a test tube (see Tay et al., 2012). The treatment reduced the expression of LRP6 as well as levels of free β-catenin. The oligomer also reduced β-catenin in primary neurons.
Liu et al. report some hints of the same mechanism in Alzheimer's. LRP6 and free β-catenin were significantly lower in postmortem brain tissue from AD patients than age-matched controls. The researchers also found that Aβ42 and Aβ40 in soluble and insoluble brain fractions negatively correlated with LRP6 expression, suggesting a link between LRP6 deficiency and Aβ production in AD.
“This study is a very important, complete piece of work, and supports the hypothesis that loss of Wnt signaling leads to AD,” said Giancarlo De Ferrari of Andres Bello University in Santiago, Chile. Ferrari's lab discovered the association between LRP6 variants and increased risk for AD (see May 2007 news story; Alarcon et al., 2013). De Ferrari and other commentators pointed out that the connection between Wnt signaling and AD has a rich history (see De Ferrari and Inestrosa, 2000; and De Ferrari et al., 2013). In 1998, work from Bruce Yankner’s lab at Harvard University revealed that pathogenic mutations in the APP processing enzyme presenilin 1 destabilize β-catenin and render neurons susceptible to Aβ (see Zhang et al., 1998). Interestingly, Yankner's group recently reported that REST, a transcription factor regulated by Wnt, turns on in the aging brain and protects neurons from damage, in part by reducing the production of Aβ. People with AD have reduced levels of this neuroprotective factor (see Mar 2014 news story). De Ferrari said he hopes the current paper will further motivate researchers to pursue the Wnt signaling pathway as a therapeutic target.—Jessica Shugart
References
News Citations
- Genetics Link Late-onset AD to Chromosome 12 and Wnt Signaling
- No REST for Weary Neurons: Protective Factor Stems Cognitive Decline
Paper Citations
- Cerpa W, Godoy JA, Alfaro I, Farías GG, Metcalfe MJ, Fuentealba R, Bonansco C, Inestrosa NC. Wnt-7a modulates the synaptic vesicle cycle and synaptic transmission in hippocampal neurons. J Biol Chem. 2008 Feb 29;283(9):5918-27. Epub 2007 Dec 20 PubMed.
- Cerpa W, Gambrill A, Inestrosa NC, Barria A. Regulation of NMDA-receptor synaptic transmission by Wnt signaling. J Neurosci. 2011 Jun 29;31(26):9466-71. PubMed.
- Inestrosa NC, Varela-Nallar L. Wnt signaling in the nervous system and in Alzheimer's disease. J Mol Cell Biol. 2014 Feb;6(1):64-74. PubMed.
- Purro SA, Galli S, Salinas PC. Dysfunction of Wnt signaling and synaptic disassembly in neurodegenerative diseases. J Mol Cell Biol. 2014 Feb;6(1):75-80. Epub 2014 Jan 20 PubMed.
- Alarcón MA, Medina MA, Hu Q, Avila ME, Bustos BI, Pérez-Palma E, Peralta A, Salazar P, Ugarte GD, Reyes AE, Martin GM, Opazo C, Moon RT, De Ferrari GV. A novel functional low-density lipoprotein receptor-related protein 6 gene alternative splice variant is associated with Alzheimer's disease. Neurobiol Aging. 2012 Dec 6; PubMed.
- Tay WM, Bryant JG, Martin PK, Nix AJ, Cusack BM, Rosenberry TL. A mass spectrometric approach for characterization of amyloid-β aggregates and identification of their post-translational modifications. Biochemistry. 2012 May 8;51(18):3759-66. PubMed.
- De Ferrari GV, Inestrosa NC. Wnt signaling function in Alzheimer's disease. Brain Res Brain Res Rev. 2000 Aug;33(1):1-12. PubMed.
- De Ferrari G, Avila M, Medina M, Pérez-Palma E, Bustos B, Alarcón M. Wnt/β-Catenin Signaling in Alzheimer's Disease. CNS Neurol Disord Drug Targets. 2013 Dec 22; PubMed.
- Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998 Oct 15;395(6703):698-702. PubMed.
External Citations
Further Reading
Papers
- De Ferrari GV, Chacón MA, Barría MI, Garrido JL, Godoy JA, Olivares G, Reyes AE, Alvarez A, Bronfman M, Inestrosa NC. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol Psychiatry. 2003 Feb;8(2):195-208. PubMed.
Primary Papers
- Liu CC, Tsai CW, Deak F, Rogers J, Penuliar M, Sung YM, Maher JN, Fu Y, Li X, Xu H, Estus S, Hoe HS, Fryer JD, Kanekiyo T, Bu G. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer's disease. Neuron. 2014 Oct 1;84(1):63-77. Epub 2014 Sep 18 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.