More on Moribund Mitochondrial Respiration Prior to Plaques
Quick Links
The brain's metabolism starts to wane decades before Alzheimer's symptoms. Why the energy deficit? Scientists led by Andrés Norambuena and George Bloom at the University of Virginia, Charlottesville, blame failure of a specific type of mitochondrial respiration called nutrient-induced mitochondrial activation. NiMA, they write in a bioRxiv preprint posted on February 4, weakens in mouse models of amyloidosis a year before plaques appear. They believe Aβ oligomers stifle mitochondria by activating GSK3β, a kinase linked to AD. “Disruption of NiMA in the brain could be a very early event in AD pathogenesis in humans,” the authors wrote.
Previously, Norambuena and Bloom described NiMA, a signaling pathway between lysosomes and mitochondria, in cultured human neurons, whereby insulin or a mixture of arginine and leucine activates the master regulatory kinase mTOR on the lysosome surface. This stirs mitochondria to ramp up oxidative phosphorylation; however, soluble Aβ oligomers blocked NiMA (Norambuena et al., 2018).
To look for this in vivo, first author Norambuena studied APPSAA knock-in mice, which express one copy of wild-type mouse amyloid precursor protein and another with a humanized Aβ sequence and three mutations, Swedish, Arctic, and Austrian, to speed plaque formation. These mice have Aβ oligomers in the brain and CSF by 4 months of age, and develop plaques at 16 months. In 2-, 4-, and 6-month-old mice, Norambuena analyzed mitochondrial metabolism and cellular oxygen consumption through a cranial window using two-photon fluorescence lifetime imaging and photoacoustic microscopy, respectively. Both methods measure changes in the intrinsic fluorescence of molecules: the former of two mitochondrial cofactors, the latter of oxygenated hemoglobin.
To stimulate NiMA, Norambuena dripped a solution of arginine and leucine onto the mouse cortex through the cranial window. Thirty minutes later, mitochondrial respiration increased in wild-type mice. The cocktail also jolted mitochondria in 2-month-old APPSAA mice but not in 4- or 6-month-olds (image below). “The simplest explanation for this observation is that the single mutated APP gene drove Aβ production to levels sufficient to block NiMA,” the authors concluded.
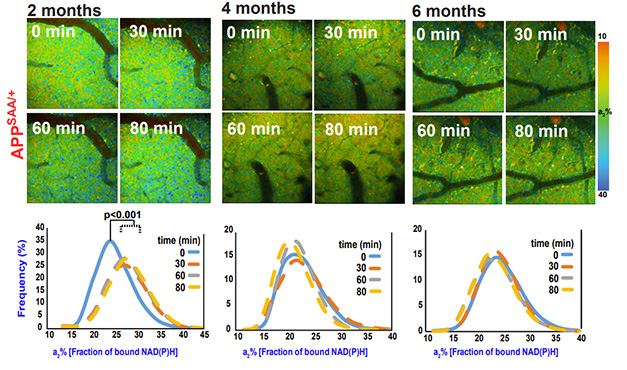
Dwindling NiMA. Through a cranial window, two-photon microscopy captured mitochondrial activity up to 80 minutes after stimulation. It rose (blue puncta) in 2-, but not 4- or 6-month-old APPSAA mice. Mitochondrial respiration was measured by the fraction of NAD(P)H bound to mitochondrial respiratory chain enzymes, a proxy for the state of oxidative phosphorylation (a2 percent). [Courtesy of Norambuena et al., bioRxiv, 2024.]
To find molecules that regulate NiMA, Norambuena screened for protein kinases, turning up GSK3β. It hyperphosphorylates tau, promotes Aβ production, and lowers mitochondrial metabolism. A GSK3β inhibitor reportedly increased mitochondrial respiration in mouse hippocampus (Nov 2016 news; Martin et al., 2018).
To see if this might rescue NiMA, Norambuena dripped the GSK3β inhibitor TWS119 through the cranial window. Mitochondrial respiration in the brains of 4-month-old wild-type mice jumped 30 percent, but only 10 percent in 4-month-old APPSAA mice. The GSK3β inhibitor tideglusib did not stimulate NiMA in wild-type mice.
The authors think trouble for NiMA results from an interplay between Aβ oligomers, GSK3β, and the metabolic regulator mTORC1. The latter controls a broad range of cellular processes ranging from protein synthesis to autophagy, and mitochondrial dynamics such as mitophagy. Previously, Bloom's group had reported that Aβ oligomers reduce lysosomal mTORC1 activation, and therefore NiMA, by inhibiting insulin signaling, and that they activate mTORC1 on the plasma membrane to nudge neurons into the cell cycle (Norambuena et al., 2022; Norambuena et al., 2017).
“It is reasonable to speculate that during AD progression, the combination of Aβ production and insulin resistance leads to an imbalance in mTORC1 activity favoring its ectopic and toxic activity at the plasma membrane over its normal physiological functions on lysosomes,” the authors wrote (image below).
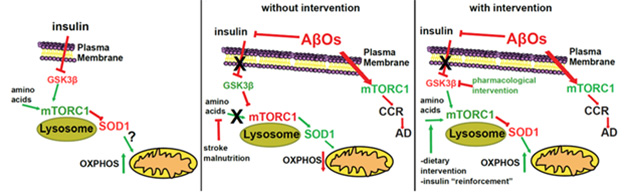
Modeling NiMA. In wild-type mice (left), insulin inhibits GSK3β, allowing amino acids to stimulate lysosomal mTORC1 and mitochondrial oxidative phosphorylation (NiMA). In AD (middle), Aβ oligomers release this brake on lysosomal mTORC1, decreasing NiMA, and activate mTORC1 ectopically on the plasma membrane, triggering cell cycle re-entry (CCR). Blocking GSK3β with a drug may restore NiMA (right). [Courtesy of Norambuena et al., bioRxiv, 2024.]
Angelika Harbauer of the Max Planck Institute of Neurobiology in Munich remains unconvinced. “I believe it is too early to claim a causal relationship between APP mutations, GSK3β, mTORC1, and mitochondrial dysfunction in neurons in AD, given the challenges in translating in vitro mechanistic analyses in isolated cell types to the complex cellular environments and interactions in vivo,” she wrote (comment below).—Chelsea Weidman Burke
References
Research Models Citations
Mutations Citations
News Citations
Therapeutics Citations
Paper Citations
- Norambuena A, Wallrabe H, Cao R, Wang DB, Silva A, Svindrych Z, Periasamy A, Hu S, Tanzi RE, Kim DY, Bloom GS. A novel lysosome-to-mitochondria signaling pathway disrupted by amyloid-β oligomers. EMBO J. 2018 Nov 15;37(22) Epub 2018 Oct 22 PubMed.
- Martin SA, Souder DC, Miller KN, Clark JP, Sagar AK, Eliceiri KW, Puglielli L, Beasley TM, Anderson RM. GSK3β Regulates Brain Energy Metabolism. Cell Rep. 2018 May 15;23(7):1922-1931.e4. PubMed.
- Norambuena A, Sun X, Wallrabe H, Cao R, Sun N, Pardo E, Shivange N, Wang DB, Post LA, Ferris HA, Hu S, Periasamy A, Bloom GS. SOD1 mediates lysosome-to-mitochondria communication and its dysregulation by amyloid-β oligomers. Neurobiol Dis. 2022 Jul;169:105737. Epub 2022 Apr 20 PubMed.
- Norambuena A, Wallrabe H, McMahon L, Silva A, Swanson E, Khan SS, Baerthlein D, Kodis E, Oddo S, Mandell JW, Bloom GS. mTOR and neuronal cell cycle reentry: How impaired brain insulin signaling promotes Alzheimer's disease. Alzheimers Dement. 2017 Feb;13(2):152-167. Epub 2016 Sep 29 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Norambuena A, Sagar VK, Wang Z, Raut P, Feng Z, Wallrabe H, Pardo E, Kim T, Alam SR, Hu S, Periasamy A, Bloom GS. Disrupted mitochondrial response to nutrients is a presymptomatic event in the cortex of the APPSAA knock-in mouse model of Alzheimer disease. 2024 Feb 04 10.1101/2024.02.02.578668 (version 1) bioRxiv.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.