Single-Cell Methylomes Offer Clues to Aging
Quick Links
A new single-cell omics model might help scientists better understand brain function. As reported in the September 24 Nature Neuroscience, researchers led by Kate Meyer, Duke University, Durham, North Carolina, profiled the mRNA methylomes of 7,702 cells from the mouse cerebral cortex. They found that transcripts are less methylated in microglia than in other cells, that methylation patterns distinguish neuronal subtypes, and that, for thousands of transcripts, including some linked to cognition or neurodegeneration, this post-transcriptional modification changes with age. Of 15 nucleotides on the APP transcript that are methylated, 14 ditch the hydrocarbon once mice reach 15 months. What that means for protein production remains to be seen.
- New mouse model enables single-cell messenger RNA methylomics.
- Modified transcripts distinguish cell types and subtypes.
- APP mRNA became hypomethylated with age.
Methylation of nitrogen 6 in the adenosine ring, or m6A, is the most common chemical change cells make to their own mRNA. The modification can be both physiological and pathological, and bulk analysis shows that it occurs in the brain more than in other tissues and organs. But if, and how, methylation patterns differ among brain cells has been difficult to tease out because it typically requires a lot of RNA for analysis.
Now, Meyer and colleagues have found a way to map m6A sites that is compatible with single-cell RNA-Seq. They constructed a chimera comprising the cytidine deaminase APOBEC1 and the YTH binding domain found in m6A “readers.” Readers are proteins that bind methylated sites to regulate mRNA translation. They form a troika with “writers,” aka methylases, and “erasers,” aka de-methylases, to control all manner of biological processes.
Expressed in cells, the reader component of this chimera found m6A sites, then the deaminase converted adjacent cytidines to uracil, which is detectable by RNA-Seq. The authors called the method DART, or deamination adjacent to RNA modification targets.
First author Matthew Tegowski and colleagues aimed this dart at mice. They engineered animals to ubiquitously express a version of APOBEC1-YTH that can be induced by giving the animal tamoxifen. Once turned on, it robustly converted cytidine-to-uracil throughout the brain. Bulk DART-Seq identified almost 18,000 methylation sites in the cortex, about 13,500 in the hippocampus, and nearly 11,400 in the cerebellum. Pairwise comparison between these regions identified up to 3,000 sites that were differentially methylated.

Missing Methyls? Among brain cells, microglia stand out for methylating far fewer RNA sites. [Courtesy of Tegowski et al., Nature Neuroscience 2024.]
Which cells might account for the difference? Tegowski turned to single-cell DART-Seq, finding 28,412 distinct RNA methylation sites among 7,702 cells in the cortex. Most of the 11 main cell types had about the same number of methylated transcripts, except microglia. They stood out as having far less, despite expressing equivalent levels of the DART machinery as did other cells (image at right). The authors confirmed the dearth of methylation in purified microglia using mass spectrometry of polyA RNA, and a chemical assay of total RNA (Liu et al., 2023).
Did cells methylate the same RNAs? For the most part, yes. Among cells of the same type, fewer than 20 transcripts were found to be differentially methylated in a cell-to-cell comparison. Things were more heterogenous when comparing different cell types, however. Compared to astrocytes, glutamatergic neurons methylated an additional 200 RNAs.
Further, there were some transcripts, particularly of immediate early genes involved in cell signaling, that were differentially methylated among neurons of different cortical layers. Clustering by methylation, rather than by transcriptome, Tegowski identified 18 distinct subtypes of glutamatergic neuron. Each had between 70 and 200 differentially methylated transcripts.
Some mRNAs differed at just one or two m6A sites, others at 10 or more. Some of those most differentially methylated play important roles in glutamatergic signaling, such as Grin2b and Gria2, which encode subunits 2b and 2 of the NMDA receptor and AMPA receptors, respectively. These m6A cluster subtypes were distinct from transcriptomics-based subtypes (image below).
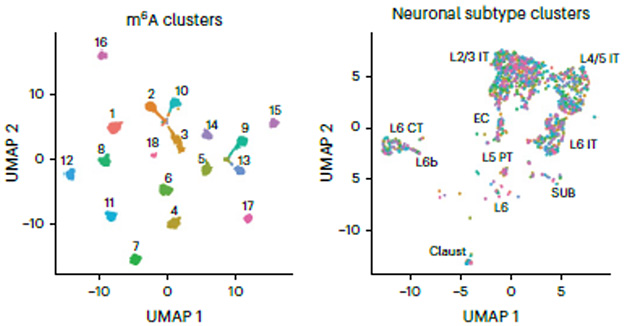
A Different Type of Subtype. DART-Seq-based cluster analysis identified 18 subtypes of glutamatergic neurons (left, color-coded). Many are represented among distinct transcript-based subtypes (right), showing that m6A clusters do not merely reflect mRNA abundance. [Courtesy of Tegowski et al., Nature Neuroscience, 2024.]
The authors also found that methylation changed with age. Comparing methylomes of 8- to 9-week-old mice with those of 14- to 15-month-olds revealed little change in some cells, including microglia, pericytes, and smooth muscle cells, but hundreds of changes in other cells, including astrocytes, oligodendrocytes, GABAergic, and glutamatergic neurons.
The latter were most affected, with methylation of more than 800 mRNAs changing with age, most being hypermethylated. Those most differentially methylated included Gria2, APP, and Lrp1, which encodes a lipoprotein receptor that has been linked to AD (Mar 2020 news). Taking a closer look at APP, the scientists found that 14 of 15 or its methylation sites lose the methyl with age.
What could all this mean for protein expression and cell biology? Curiously, the authors found no link between methylation status and transcript levels. This was true across the different glutamatergic neuron subtypes, and for the transcripts that were most heavily or hyper- or hypomethylated, such as Grin2b in glutamatergic subtypes or APP in older mice. The authors think that the main role of methylation may not be to control RNA stability or abundance.
That said, this modification has been shown to control translation. “It is possible that m6A-mediated regulation of protein production or RNA localization helps drive functional outputs of the cellular heterogeneity we observed,” the authors noted. They also posit that methylation might affect APP in a pathological context or that the hypomethylation seen in microglia might change if these cells are activated. “… [I]t will be interesting to profile m6A methylation in the aged brain from Alzheimer's disease or other disease models and investigate whether differential methylation of App and other disease-associated transcripts impacts their expression,” they wrote.—Tom Fagan
References
News Citations
Paper Citations
- Liu C, Sun H, Yi Y, Shen W, Li K, Xiao Y, Li F, Li Y, Hou Y, Lu B, Liu W, Meng H, Peng J, Yi C, Wang J. Absolute quantification of single-base m6A methylation in the mammalian transcriptome using GLORI. Nat Biotechnol. 2023 Mar;41(3):355-366. Epub 2022 Oct 27 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Tegowski M, Prater AK, Holley CL, Meyer KD. Single-cell m6A profiling in the mouse brain uncovers cell type-specific RNA methylomes and age-dependent differential methylation. Nat Neurosci. 2024 Sep 24; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.