Reelin Variant Wards Off Dementia in Colombian Kindred Siblings
Quick Links
A Colombian brother and sister staved off autosomal-dominant Alzheimer’s disease for 20 years past the age of onset expected based on their inheritance of the E280A Paisa presenilin mutation, according to a new case report. That is thanks to a rare variant in Reelin, an extracellular matrix protein that binds to the ApoE receptor, say Yakeel Quiroz at Massachusetts General Hospital and Joseph Arboleda-Velasquez of Massachusetts Eye and Ear, both in Boston; Diego Sepulveda-Falla of the University Medical Center Hamburg-Eppendorf, Germany; and Francisco Lopera of the University of Antioquia, Medellín, Colombia.
- Man carrying both the H3447R Reelin and E280A PS variants remained dementia-free 20 years after expected symptom onset.
- Brain amyloid load was high, entorhinal neurofibrillary tangles sparse.
- The variant boosts Reelin signaling, quelling tau phosphorylation.
Joachim Herz of the University of Texas Southwestern Medical Center, Dallas, who has studied Reelin in the brain for decades, was unsurprised that a variant was protective, though the magnitude of protection in the siblings struck him. “I was shocked that it could stave off cognitive decline for 20 to 30 years in someone with such a strong mutation like Paisa PSEN1,” he told Alzforum.
These researchers previously discovered a protective APOE3 variant in a woman from the same Colombian cohort. In the May 15 Nature Medicine, they report that the brother had a high amyloid load, yet fewer tau tangles, especially in his entorhinal cortex, than expected upon his death. The Reelin variant both he and his sister carried, dubbed COLBOS for the Colombian-Boston collaboration, was more active than the wild-type, modulating downstream kinases to hinder tau phosphorylation. His sister also seemed protected, though a history of brain trauma complicates her case. Exactly how this variant protects might inform the search for new therapeutics.
“It is really exciting to have a second protective variant to understand why some people are resilient to such an aggressive form of AD,” Guojun Bu, SciNeuro Pharmaceuticals, Rockville, Maryland, told Alzforum. Jason Ulrich, Washington University in St. Louis, agreed that protective genetic factors in ADAD carriers can generate new insight into how amyloid pathology can facilitate the onset of tau aggregation and subsequent neurodegeneration (comment below).
Four years ago, the case of a woman carrying the APOE3 Christchurch variant captivated researchers’ attention. Despite carrying the E280A presenilin 1 mutation, which triggers massive amyloid accumulation, neurodegeneration, and cognitive decline by age 45, she stayed cognitively intact until her 70s (Nov 2019 news; Sep 2022 news). Since then, Quiroz and colleagues have been searching for others “escapees,” identifying the brother and sister with uncharacteristic resilience to dementia.
The man remained sharp until age 70, when he was diagnosed with mild cognitive impairment. He had mild dementia by age 72 and died two years later of pneumonia. His sister's memory began slipping at age 58; she had severe dementia upon clinical evaluation at age 64 and died at age 73. However, she also had multiple co-morbidities, including a severe head injury after a fall, hypertension, and depression, all possibly contributing to earlier dementia onset than her brother.

Resilient Reelin. PET scans of the man with both the Paisa PSEN1 mutation and the H3447R Reelin variant (left) show more amyloid plaques (top) yet fewer neurofibrillary tangles (bottom) than does a typical Paisa carrier with early dementia (right). [Courtesy of Lopera et al., Nature Medicine, 2023.]
Shortly after his dementia diagnosis, the brother traveled to Boston for amyloid and tau PET scans. He had 48 percent more amyloid plaques than do typical Paisa carriers with early dementia, yet his global tau tangle burden was low (image below). Tangles were sparse in his entorhinal cortex, where tau drastically accumulates in Paisa mutation carriers. This likely spared neurons because, upon postmortem analysis, the researchers counted a higher neuron density in his entorhinal cortex than in others in this cohort. “This is key to protecting against cognitive impairment, despite the continued accumulation of tau and Aβ in the rest of the brain,” noted Inmaculada Cuchillo Ibañez of Spain’s Instituto de Neurociencias (comment below).
To compare this man’s brain to that of the previously identified APOE3 Christchurch carrier, co-first author Nelson David Villalba-Moreno of UMC Hamburg-Eppendorf looked at their autopsies. The reelin case had a similar amyloid burden, more phospho-tau, and microglia that were less activated, as determined by their size and circularity, than did the Christchurch case (see image below). Both had distinctly less ApoE staining inside their cortical and hippocampal neurons than did others in this kindred who had died in their 50s or 60s.
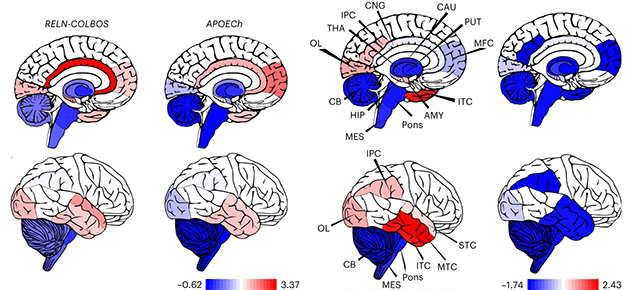
Reelin versus Christchurch. While amyloid pathology was similar in Reelin-COLBOS and ApoE Christchurch carriers (left), the former had more phospho-tau (right). [Courtesy of Lopera et al., Nature Medicine, 2023.]
How might this variant protect? Mouse amyloidosis models that lack Reelin more quickly accumulate plaques, tangles, which are unusual in amyloidosis models, and learning and memory problems, suggesting Reelin protects neurons from AD (Jul 2015 news; Jul 2010 news). A recent genome-wide association study linked a mutation in Dab 1, a kinase downstream of Reelin, to faster AD progression in APOE4 carriers, suggesting that the Reelin-Dab1 pathway helps stave off late-onset AD, too (Bracher-Smith et al., 2022).
Indeed, the H3447R variant ramped up Reelin signaling, as indicated by high levels of phosphorylated Dab1 in primary mouse cortical neurons. Likewise, 6- to 12-month-old mice into which two copies of this variant had been knocked had more phosphorylated Dab1 and more Reelin oligomers, both signs of activated signaling, in their cerebella than did wild-type animals. In the adult brain, expression in the cerebellum is highest. In the context of tauopathy, 18-month-old crosses of the knock-ins with P301L tauopathy mice had 30 percent less p-tau205 in their hippocampi and showed no sign of their typical hindlimb paralysis.
“It’s surprising that a point mutation increases Reelin activity and that such a small difference affects in vivo phenotype,” wrote Mitsuharu Hattori and Takao Kohno from Nagoya City University, Japan (comment below).
The location of the H3447R mutation, near a heparin-binding site in Reelin, might inform potential treatment strategies. The Christchurch mutation also falls within a heparin-binding area of ApoE3 and influences how it interacts with other glycosaminoglycans. GAGs act as scaffolds for signaling ligands, drawing them near receptors to drive binding and boost signaling. “Both the Christchurch and Reelin variants converge on GAG pathways as a mediator of Aβ-related tau pathology, suggesting GAGs as a potential drug target,” said Michael Ewers, Ludwig Maximilian University in Munich.—Chelsea Weidman Burke
References
Mutations Citations
News Citations
- Can an ApoE Mutation Halt Alzheimer’s Disease?
- In Brain With Christchurch Mutation, More ApoE3 Means Fewer Tangles
- Reelin in Aβ Damage?
- Research Brief: Reelin Loss Ramps Up Aβ Pathology
Research Models Citations
Paper Citations
- Bracher-Smith M, Leonenko G, Baker E, Crawford K, Graham AC, Salih DA, Howell BW, Hardy J, Escott-Price V. Whole genome analysis in APOE4 homozygotes identifies the DAB1-RELN pathway in Alzheimer's disease pathogenesis. Neurobiol Aging. 2022 Nov;119:67-76. Epub 2022 Jul 29 PubMed.
Further Reading
Papers
- Herz J, Chen Y. Reelin, lipoprotein receptors and synaptic plasticity. Nat Rev Neurosci. 2006 Nov;7(11):850-9. PubMed.
Primary Papers
- Lopera F, Marino C, Chandrahas AS, O'Hare M, Villalba-Moreno ND, Aguillon D, Baena A, Sanchez JS, Vila-Castelar C, Ramirez Gomez L, Chmielewska N, Oliveira GM, Littau JL, Hartmann K, Park K, Krasemann S, Glatzel M, Schoemaker D, Gonzalez-Buendia L, Delgado-Tirado S, Arevalo-Alquichire S, Saez-Torres KL, Amarnani D, Kim LA, Mazzarino RC, Gordon H, Bocanegra Y, Villegas A, Gai X, Bootwalla M, Ji J, Shen L, Kosik KS, Su Y, Chen Y, Schultz A, Sperling RA, Johnson K, Reiman EM, Sepulveda-Falla D, Arboleda-Velasquez JF, Quiroz YT. Resilience to autosomal dominant Alzheimer's disease in a Reelin-COLBOS heterozygous man. Nat Med. 2023 May;29(5):1243-1252. Epub 2023 May 15 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University
This is another exciting discovery from the Colombia-Boston collaboration identifying potential protective genetic factors in ADAD carriers. These unique genetic examples provide valuable insight into new biology regarding how amyloid pathology can facilitate the onset of tau aggregation and subsequent neurodegeneration. I was intrigued by the data showing the COLBOS variant in Reelin increased binding to HSPGs. Some studies have implicated tau binding to HSPGs as an important route of entry for transcellular spreading of pathological tau conformations, so it is interesting that variants in Reelin and APOE that affect the affinity of the respective proteins for HSPGs potentially protect against the development or spread of tau pathology (Arboleda-Velasquez et al., 2019; Holmes et al., 2013; Rauch et al., 2018).
The previously identified APOE-Christchurch variant decreased the affinity of APOE for HSPGs, raising the question of whether differential APOE vs. Reelin binding to HSPGs, and potentially VLDLR or APOER2, regulate tau spreading. In addition, Reelin-Dab1 signaling, which is evidently elevated with the COLBOS variant, has previously been shown to repress tau phosphorylation (Hiesberger et al., 1999). This raises the question of whether Dab1 signaling is upregulated with APOE-Christchurch expression, although it is entirely possible that APOE-Christchurch and Reelin-COLBOS repress tau pathology via less directly related mechanisms. Hopefully emerging studies in iPSC organoids or new APOE-Christchurch KI mice can address this potential functional interaction.
Finally, it is worth noting that a recent GWAS report found SNPs in DAB1 that increased disease risk only in APOE4-homozygous individuals, which the authors speculated may suggest a particular role for RELN-DAB1 signaling in regulating tau pathology in the context of high levels of amyloid pathology, such as would exist in ADAD (Bracher-Smith et al., 2022).
References:
Arboleda-Velasquez JF, Lopera F, O'Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM, Quiroz YT. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680-1683. Epub 2019 Nov 4 PubMed.
Holmes BB, DeVos SL, Kfoury N, Li M, Jacks R, Yanamandra K, Ouidja MO, Brodsky FM, Marasa J, Bagchi DP, Kotzbauer PT, Miller TM, Papy-Garcia D, Diamond MI. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013 Aug 13;110(33):E3138-47. Epub 2013 Jul 29 PubMed.
Rauch JN, Chen JJ, Sorum AW, Miller GM, Sharf T, See SK, Hsieh-Wilson LC, Kampmann M, Kosik KS. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci Rep. 2018 Apr 23;8(1):6382. PubMed.
Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999 Oct;24(2):481-9. PubMed.
Bracher-Smith M, Leonenko G, Baker E, Crawford K, Graham AC, Salih DA, Howell BW, Hardy J, Escott-Price V. Whole genome analysis in APOE4 homozygotes identifies the DAB1-RELN pathway in Alzheimer's disease pathogenesis. Neurobiol Aging. 2022 Nov;119:67-76. Epub 2022 Jul 29 PubMed.
Instituto de Neurociencias, Universidad Miguel Hernandez
This extensive and comprehensive article includes a huge variety of methodological approaches both in vivo and in vitro to explore the effect of the RELN-COLBOS mutation on the reelin signaling pathway. To further include the data on the total levels of Dab1 (not only phosphorylated Dab1), as well as more experiments on the effect of the mutant reelin protein on tau and phospho-tau in mouse cortical neurons (if they are not in the supplementary figures), would have positively completed the article, if possible!
Our laboratory, i.e. Javier Saez-Valero's lab, has extensively studied reelin protein in human tissues from individuals with Alzheimer's. We have found certain alterations that impair its protective function, including the ability to bind to its receptor ApoER2 and its ability to control tau phosphorylation.
The present article highlights that the RELN-COBOS mutation has a greater beneficial effect on tau accumulation in the entorhinal region of the brain. This is very interesting because it is the region where the progressive accumulation of tau tangles in Alzheimer's disease begins, which suggests this fact is key to protect against cognitive impairment, despite the continued accumulation of tau and Aβ in the rest of the brain.
The comparison of RELN-COLBOS and APOEChr is also very interesting. Both mutations protect against Alzheimer's, and although the effects on the accumulation of tau and Aβ are not the same on the individuals who carry the mutation, reelin and ApoE bind to the same receptor, ApoER2, indicating that this receptor and the signaling cascade initiated upon its activation may be key in Alzheimer's disease and deserve more investigation.
Nagoya City University
Nagoya City University
This is the first causal link between Reelin activity and AD, and the first gain-of-function Reelin mutation in humans. That a point mutation in Reelin’s C-terminal region increases activity is surprising. It is even more surprising that such a small difference in Reelin affects the in vivo phenotype. We previously showed that the C-terminal region of Reelin is necessary for efficient activation of its downstream signaling and that this effect is mediated, at least in part, by Nrp1. Their findings matches our story and reinforces it.
Previous animal studies suggest Reelin beneficial action in AD, but there is no conclusive evidence in humans. Here the newly found Reelin missense mutation in a PSEN1-mutant man seems to protect him from getting AD. If this is the case, upregulating Reelin activity will be beneficial to AD patients.
The authors found that the Reelin H3447R mutation in a man with PSEN1 mutation had delayed the onset of cognitive decline and reduced tau pathology. Furthermore, they found that this mutation dramatically increased Reelin binding to glycosaminoglycans and Neuropilin-1 (Nrp1).
Nrp1 is a co-receptor of Reelin and enhances Reelin-binding with the canonical receptor (Kohno et al., 2020). The interaction between Reelin and Nrp1 is dependent on the C-terminal region of Reelin, which includes H3447. Therefore, the observation that H3447R mutation stabilizes the interaction between Reelin and Nrp1 makes sense.
A previous work (Kocherhans et al., 2010) suggested that Reelin inhibits Aβ production. Another (Pujadas et al., 2014) suggested that Reelin binds Aβ and decreases its toxicity. These mechanisms can be compatible with the one proposed by Lopera and colleagues. Boosting Reelin function or upregulating its production or secretion may be a promising strategy.
References:
Kohno T, Ishii K, Hirota Y, Honda T, Makino M, Kawasaki T, Nakajima K, Hattori M. Reelin-Nrp1 Interaction Regulates Neocortical Dendrite Development in a Context-Specific Manner. J Neurosci. 2020 Oct 21;40(43):8248-8261. Epub 2020 Oct 2 PubMed.
Kocherhans S, Madhusudan A, Doehner J, Breu KS, Nitsch RM, Fritschy JM, Knuesel I. Reduced Reelin expression accelerates amyloid-beta plaque formation and tau pathology in transgenic Alzheimer's disease mice. J Neurosci. 2010 Jul 7;30(27):9228-40. PubMed.
Pujadas L, Rossi D, Andrés R, Teixeira CM, Serra-Vidal B, Parcerisas A, Maldonado R, Giralt E, Carulla N, Soriano E. Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer's disease. Nat Commun. 2014 Mar 6;5:3443. PubMed.
Rouen University Hospital
Looking for modifiers of age of clinical onset in PSEN1-mutation carriers is a very interesting and important aim, as it helps us better understand AD pathophysiology and could point toward actionable targets for disease modification. This task is difficult for two main reasons. There are many distinct mutations in PSEN1 (as well as in PSEN2 and, to a lesser extent, APP). Per mutation, there is limited variance of ages of onset (AOO). This is contrary to frontotemporal dementia, where, for example C9ORF72 and GRN mutations have a wide range of AOO within and between families.
Here the authors took advantage of two unique opportunities: the very large size of the well-known kindred with carriers of the PSEN1 E280A mutation and existence of a few outliers beyond the known AOO variance. In this kindred, the effect of APOE genotypes had previously been assessed as possible contributors to AOO variance in mutation carriers. A role for APOE4 was first suggested to lower AOO (Pastor et al., 2003); which was not confirmed in another paper where APOE4 had no effect but APOE2 delayed AOO (Pastor et al., 2003). Authors from the same group as the current paper previously reported a homozygous variant in APOE, called Christchurch, as a likely explanation of unexpected AOO in one outlier from this kindred (Arboleda-Velasquez et al., 2019).
In this new study, a rare RELN variant is proposed to participate in delayed AOO in two sibpairs with outlier AOO as well, who do not carry the Christchurch APOE variant. The paper provides convincing evidence, in vitro and in vivo, that this RELN variant is not neutral in terms of protein function. Reelin has been studied for years as a modifier of Aβ peptide aggregation and as a driver, through its receptors ApoeR2 and/or VLDLR and transducer Dab1, of tau phosphorylation. Here, the observation of a high amyloid load with relatively lower tau burden on PET in carriers of both the PSEN1 mutation and the rare RELN variant (or the APOE Christchurch homozygous carrier) led the authors to hypothesize that the variant may relatively protect against tau pathology. Although tau burden remained in similar ranges as in younger MCI PSEN1 mutation carriers from the family, comparisons of single observations at different ages remain challenging. The authors provide evidence that the variant is associated with increased Dab1 phosphorylation, increased interactions with heparin and reduced tau phosphorylation, although the effects in knock-in mice do not show suggestive changes in females or in heterozygous males.
This observation is very interesting. As the authors acknowledge, it is challenging to definitively confirm that this variant is responsible for the delayed onset in both carriers. Genetics arguments are indeed rather limited here. The RELN variant is extremely rare and no recurrence was observed in the pedigree or in other families. This could be considered to argue in favor of a role of this variant in the carriers, given the rarity of AOO outliers in the pedigree, but one could also argue that this could be due to the rarity of the variant itself, knowing that every individual carries a large burden and diversity of rare variants overall.
Importantly, transmission of this variant is unknown. If it was inherited from the mother with early onset dementia, this would argue against a role. This hypothesis is 50 percent likely, as the mother herself could have inherited the variant by chance from her parent unrelated to the rest of the PSEN1 family (explaining that it is not found in other individuals of this family). If inherited from the father (and if he is not related to the PSEN1 kindred), it would remain consistent with a possible effect on delaying AOO, but we cannot exclude a coincidence either. Haplotype analyses might help determine parental transmission, based on DNA available in relatives.
The filtration and prioritization of the variants here is presented as the way researchers came to study the RELN variant. This strategy can be risky, because it was performed using a single individual genome with a single algorithm (exomiser/genomizer). This puts weights on genes and variants using arguments that could drastically change the prioritization, should the gene lists and different thresholds be a little bit modified. For example, most noncoding regions of genes (introns, UTRs), seem to have been removed from the simplex whole genome sequencing analysis, although the authors mention an APP 3'UTR variant, which seems to have been considered following a closer look at a list of AD-linked genes (this list being otherwise nonconsensual). Anyway, there is no good or bad way to prioritize variants based on a single individual. It is already a difficult task when dealing with variant interpretation for Mendelian diseases in a clinical setting, and even more difficult when looking for a putative modifier in this context in a single case. It is like searching a needle in a haystack without being sure that there is any needle or a single needle to be found. Such a prioritization is thus not evidence by itself, I see it more as a help to search for more genetic evidence on a short list of variants, starting from a huge list of individual genomic variants.
Here, the authors provide compelling evidence of a non-neutral effect of this variant on Reelin function, acting as a likely hypermorph and thus consistent with the hypothesis of protection against Aβ-induced toxicity. That said, some evidence is missing that this variant drives the AOO effect, precluding use of genetic counseling of the carriers' children, for example. The authors discuss this. I agree with them that these results should be interpreted with caution while they can help generate hypotheses and further research on the topic.
References:
Pastor P, Roe CM, Villegas A, Bedoya G, Chakraverty S, García G, Tirado V, Norton J, Ríos S, Martínez M, Kosik KS, Lopera F, Goate AM. Apolipoprotein Eepsilon4 modifies Alzheimer's disease onset in an E280A PS1 kindred. Ann Neurol. 2003 Aug;54(2):163-9. PubMed.
Arboleda-Velasquez JF, Lopera F, O'Hare M, Delgado-Tirado S, Marino C, Chmielewska N, Saez-Torres KL, Amarnani D, Schultz AP, Sperling RA, Leyton-Cifuentes D, Chen K, Baena A, Aguillon D, Rios-Romenets S, Giraldo M, Guzmán-Vélez E, Norton DJ, Pardilla-Delgado E, Artola A, Sanchez JS, Acosta-Uribe J, Lalli M, Kosik KS, Huentelman MJ, Zetterberg H, Blennow K, Reiman RA, Luo J, Chen Y, Thiyyagura P, Su Y, Jun GR, Naymik M, Gai X, Bootwalla M, Ji J, Shen L, Miller JB, Kim LA, Tariot PN, Johnson KA, Reiman EM, Quiroz YT. Resistance to autosomal dominant Alzheimer's disease in an APOE3 Christchurch homozygote: a case report. Nat Med. 2019 Nov;25(11):1680-1683. Epub 2019 Nov 4 PubMed.
Barcelona Supercomputing Center
Lopera and colleagues showcase the importance of studying rare mutations in large, well-characterized populations as the Colombian ADAD cohort.
Three points caught my attention. First, the sex differences between the siblings and also in animal models. A lot of literature has demonstrated sex-specific pathophysiological changes (Ferretti et al., 2018), and it’s intriguing that this Reelin-based mechanism described in their paper is also sex-dependent. Second, it's important to notice the similarity on the protein-binding profile of the mutated regions for both RELN-COLBOS and the ApoECh variants. In both cases, the mutation is in the heparin-binding area, and they both interact with glycosaminoglycans. In this sense, recent data on a pathological increase in ApoE glycosylation (Lawler et al., 2023) might point to age-related glycosylation in the etiology of the disease.
Finally, the presence of the RELN-COLBOS mutation not only increases postmortem CTERM staining of RELN, but also led to lower APOE findings. This decrease might reflect a lower neuroinflammatory profile in the RELN-COLBOS mutation compared to sAD and ADAD, a point worth further exploration.
The ApoECh and the RELN-COLBOS cases point to novel pharmacological alternatives beyond amyloid and tau, but further study is needed (Nov 2019 news) to evaluate if drugs that target ApoE- and/or Reelin-related mechanisms might work as disease-modifying therapies (Ferretti et al., 2018), study the dynamics of ApoE and Reelin, and interaction with their competitive receptors along the disease (Lawler et al., 2023) and pay extra attention to the sex effect.
References:
Ferretti MT, Iulita MF, Cavedo E, Chiesa PA, Schumacher Dimech A, Santuccione Chadha A, Baracchi F, Girouard H, Misoch S, Giacobini E, Depypere H, Hampel H, Women’s Brain Project and the Alzheimer Precision Medicine Initiative. Sex differences in Alzheimer disease - the gateway to precision medicine. Nat Rev Neurol. 2018 Aug;14(8):457-469. PubMed.
Lawler PE, Bollinger JG, Schindler SE, Hodge CR, Iglesias NJ, Krishnan V, Coulton JB, Li Y, Holtzman DM, Bateman RJ. Apolipoprotein E O-glycosylation is associated with amyloid plaques and APOE genotype. Anal Biochem. 2023 Jul 1;672:115156. Epub 2023 Apr 16 PubMed.
SUNY Upstate Medical University
The implication that a gain-of-function mutation in RELN is involved in the protection from autosomal-dominant AD in the Lopera et al. study is very exciting, since it could provide a path to a pharmacological intervention that blocks AD presentation even in the presence of a high Aβ plaque load. The proposal that this may work through a competition between REELIN and APOE3 for binding to the REELIN receptors that are also APOE receptors is intriguing, and is supported by other work in the field. Importantly, this suggests a dark side to APOE3, and provides evidence that this dark side has to do with the suppression of REELIN signaling.
The Bracher-Smith et al. study (2022) also connected the REELIN pathway, APOE, and AD. In this study, however, the genetic effect of DAB1, which encodes an obligate signaling adaptor on the pathway, was only consequential in APOE4 homozygotes in late-onset AD, while in the Lopera et al. study APOE3 alleles are present. Thus more work is needed to fully understand the context of when REELIN pathway manipulation may be beneficial to patients.
References:
Bracher-Smith M, Leonenko G, Baker E, Crawford K, Graham AC, Salih DA, Howell BW, Hardy J, Escott-Price V. Whole genome analysis in APOE4 homozygotes identifies the DAB1-RELN pathway in Alzheimer's disease pathogenesis. Neurobiol Aging. 2022 Nov;119:67-76. Epub 2022 Jul 29 PubMed.
HudsonAlpha Institute for Biotechnology
This report from the COLBOS team is another important step in understanding what age-of-onset outliers in dominant AD can teach us. I would like to echo the comments of Dr. Gael Nicolas on the proposed genetic association. In general, investigation of genetic contributors to resistance to dominant AD is a difficult task. Rare variants will have the largest effect size, but therefore will also likely lack replication given the small size of dominant AD cohorts. The authors appropriately address this limitation in the text of the paper by both acknowledging that other variants could contribute to the outlier phenotype and also providing detailed characterization of the implicated RELN variant. I am hopeful that additional protective variants in RELN could be identified in future studies to strengthen this genetic association, which could mirror the identification of additional rare variants in APOE supportive of the effect of the APOE Christchurch variant.
While rare variants are the most likely to have a strong effect, it is also possible that common variants (or variants that are relatively rare, but are at a frequency that allows evaluation of replication) could contribute to resistance. We recently worked with Dr. Lopera's team to assess the association of such variants comprehensively in the 340 individuals in the E280A kindred that had reached a dementia diagnosis and had DNA available (Cochran et al., 2023). We assessed replication in two dominant AD cohorts, DIAN and ADDS, as well as in sporadic EOAD and several LOAD studies. While the associations we identified should be interpreted with caution, given the small size of the cohort, we did propose several nominal associations of interest, including several implicating clusterin. We also observed a clear two-year shift earlier in age of onset with APOE ε4 and a correlation of higher LOAD polygenic risk score with earlier age of onset in this kindred. Summary statistics from this study are open access through NHGRI-EBI.
In summary, while continued analysis of the possible contribution of rare variants is a critically important effort, it is also important to consider the possible contribution of more common genetic variation to resistance phenotypes.
References:
Cochran JN, Acosta-Uribe J, Esposito BT, Madrigal L, Aguillón D, Giraldo MM, Taylor JW, Bradley J, Fulton-Howard B, Andrews SJ, Acosta-Baena N, Alzate D, Garcia GP, Piedrahita F, Lopez HE, Anderson AG, Rodriguez-Nunez I, Roberts K, Dominantly Inherited Alzheimer Network, Absher D, Myers RM, Beecham GW, Reitz C, Rizzardi LF, Fernandez MV, Goate AM, Cruchaga C, Renton AE, Lopera F, Kosik KS. Genetic associations with age at dementia onset in the PSEN1 E280A Colombian kindred. Alzheimers Dement. 2023 Sep;19(9):3835-3847. Epub 2023 Mar 23 PubMed.
Make a Comment
To make a comment you must login or register.