Protective Microglial Gene Variant Promotes Phagocytosis
Quick Links
The immune system has recently seized the spotlight in Alzheimer’s research, with several genome-wide association studies (GWAS) linking the disease to genetic variation in microglia genes. However, it remains unclear how these genes influence pathology. Most of the genetic variants uncovered lie far from coding elements, and geneticists believe they are inherited with other, as yet unidentified polymorphisms that are the real risk factors. In the August 14 Journal of Neuroscience, researchers led by Steven Estus at the University of Kentucky, Lexington, revealed the functional variant for one of these risk genes, CD33. Previously, several GWAS identified a single nucleotide polymorphism (SNP) in the promoter region of this gene that lowers AD risk (see related ARF news). Now, Estus and colleagues link this SNP to a co-inherited one that disrupts CD33 mRNA splicing and disables the protein. Because CD33 signaling inhibits phagocytosis, the inactive form likely enhances the ability of microglia to chew up Aβ deposits, thus protecting against AD, the authors suggest. CD33 encodes an immunoglobulin-like cell-surface receptor, currently number eight on AlzGene’s Top Ten list.
The findings dovetail with recent studies that reported a link between CD33 expression and phagocytosis. Rudy Tanzi, Ana Griciuc and colleagues at Massachusetts General Hospital, Charlestown, found that microglia in the AD brain make too much of the receptor and struggle to degrade Aβ (see ARF webinar and Griciuc et al., 2013). Likewise, researchers at Brigham and Women’s Hospital led by Philip De Jager tied the CD33 risk allele to greater expression of the receptor on the surface of microglia, and less internalization of Aβ (see Bradshaw et al., 2013). The data reinforce each other and support the idea that microglia play a key role in keeping amyloid pathology in check and lowering AD risk, Estus told Alzforum. The findings also gibe with the 2012 identification of a variant in the microglial gene TREM2 as a major AD risk factor (see ARF related news story). TREM2 enhances phagocytosis, and the AD risk allele inactivates the protein. CD33 and TREM2 thus have opposing effects on microglial activation (see figure below). In both cases, mutations that suppress phagocytosis increase AD risk.
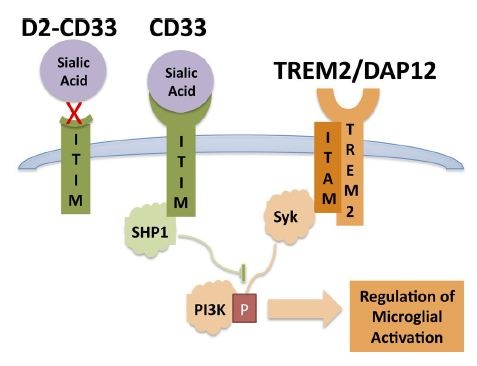
Microglial Balancing Act: By activating or blocking phosphatidyl-inositol-3 kinase (PI3K), CD33 and TREM2 have opposing actions on activation of microglia and phagocytosis of Aβ.
Image courtesy Malik et al., and the Journal of Neuroscience, 2013
Although these previous studies hinted at the mechanism by which CD33 might affect AD risk, they did not identify the functional mutation. To pin this down, first author Manasi Malik looked to different CD33 mRNA isoforms. She examined RNA isolated from 28 postmortem human AD brains and 27 age-matched controls. Many transcripts from both samples lacked exon 2, which encodes the domain that binds sialic acids. Since sialic acids activate CD33, transcripts missing exon 2 (D2-CD33) are predicted to remain quiescent. Importantly, Malik and colleagues found that while microglia from AD brains made more total CD33 than did cells from control brains, they made less D2-CD33. This suggested that the functional protective mutation might affect splicing, favoring more of the silent receptor.
What is the functional variant? The authors sequenced the CD33 gene to look for polymorphisms that co-inherit with the GWAS SNP, rs3865444. They found a second SNP that fit the bill, rs12459419, in the fourth base of exon 2. People with a T nucleotide at this locus had a higher ratio of D2-CD33 to total CD33 compared to those who carried a C at this position. In cell culture, microglia transfected with the protective T allele made three times more D2-CD33 than did those without, confirming that this SNP directly affects RNA splicing. In ongoing work, Estus and colleagues are investigating whether other splice isoforms are affected by the protective rs12459419 variant. They are also interested in using the methods in this study to go after other GWAS hits.
“The authors have designed a clever way to assess the role of one of these [GWAS] variants… and provide a functional SNP linked with the GWAS SNP,” Rita Guerreiro at University College London, U.K., wrote to Alzforum (see full comment below). In future work it will be important to test the new rs12459419 SNP in large case-control cohorts, as well as to directly test the role of sialic acids in this pathway, she suggested.
Estus noted that his study, along with those by Griciuc et al. and Bradshaw et al., suggest that inhibiting CD33 could be a therapeutic strategy for lowering amyloid pathology and delaying AD. Antibodies that repress the receptor exist, and have been tested as treatments for acute myeloid leukemia (see, e.g., this Phase 1 trial of lintuzumab). The Food and Drug Administration approved Pfizer’s anti-CD33 antibody Mylotarg for cancer treatment, but the company later voluntarily withdrew the drug from the U.S. market due to safety concerns (see FDA press release). To delay Alzheimer’s disease, a drug that stimulated phagocytosis might have to be given for many years. For this reason, a better strategy might be to develop small molecule inhibitors of CD33, Estus suggested.—Madolyn Bowman Rogers
References
News Citations
Webinar Citations
Paper Citations
- Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013 May 22;78(4):631-43. PubMed.
- Bradshaw EM, Chibnik LB, Keenan BT, Ottoboni L, Raj T, Tang A, Rosenkrantz LL, Imboywa S, Lee M, Von Korff A, , Morris MC, Evans DA, Johnson K, Sperling RA, Schneider JA, Bennett DA, De Jager PL. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013 Jul;16(7):848-50. PubMed.
External Citations
Further Reading
News
Primary Papers
- Malik M, Simpson JF, Parikh I, Wilfred BR, Fardo DW, Nelson PT, Estus S. CD33 Alzheimer's Risk-Altering Polymorphism, CD33 Expression, and Exon 2 Splicing. J Neurosci. 2013 Aug 14;33(33):13320-5. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Van Andel Institute
Even though the mechanism proposed by the authors is just a model (no experiments were done to actually demonstrate the involvement of sialic acids in the pathway), the identification of a potential functional polymorphism in linkage disequilibrium with a single nucleotide polymorphism (SNP) identified by genome-wide association studies(GWAS) is of great interest.
By design, the probability of GWAS identifying the real variant associated with a disease risk is very small. On top of this, a large proportion of the identified SNPs are located in gene deserts, non-coding gene regions, or are intergenic. This indicates that a large part of the genetic risk for common diseases is unrelated to coding variations and most probably comes from gene expression changes.
The authors have designed a clever way to assess the role of one of these variants and they show that not only is CD33 expression increased in AD, but they provide a functional SNP linked with the GWAS SNP that is involved with differential splicing of exon 2 (which encodes the IgV domain).
These and other recent results clearly show that the latest International Genomics of Alzheimer’s Project CD33 risk association needs to be re-evaluated and, once again, puts microglial pathways in the spot for Alzheimer's disease.
It will definitely be important to test rs12459419 in big cohorts of cases and controls.
View all comments by Rita GuerreiroMassachusetts General Hospital and Harvard Medical School
The article by Malik and colleagues offers new insights into the genetics of CD33, a gene that we first reported as a risk factor for Alzheimer’s disease (AD) in 2008 (Bertram et al., 2008). CD33 is a sialic acid-binding protein that we recently found to be expressed in microglial cells in the aging human brain (Griciuc et al., 2013). There, it inhibits the clearance of amyloid β, and promotes the deposition of Aβ in plaques, which are major pathological hallmarks of AD. We have previously shown that the CD33 variant, rs3865444, which protects against AD is associated with reduced CD33 microglial expression. Our finding that CD33 inactivation in mice leads to reduced amyloid pathology in the brain further suggests a critical role for CD33 in AD pathogenesis. However, the molecular mechanisms underlying the protective effect of the rs3865444 mutation has remained hidden.
In their article, Malik et al. characterize another CD33 variant, the missense substitution, rs12459419, that is in close proximity to the rs3865444 mutation; these two mutations are co-inherited. The authors suggest that rs12459419 promotes the splicing of CD33 RNA into an alternative mRNA version which lacks the sialic acid binding domain. As such, the truncated CD33 protein would be unable to carry out its cellular functions that are regulated by sialic acid binding.
Our recent study suggested a critical role for the sialic acid-binding domain of CD33 based on its inhibitory effect on amyloid beta uptake and clearance by microglia; a genetically-engineered mutant CD33 version lacking the sialic acid-binding domain is no longer capable of inhibiting amyloid beta clearance (Griciuc et al., 2013). These results are consistent with those of Malik et al. showing that the rs12459419 variant may confer protection against AD by promoting the production of a functionally defective CD33 variant in the human brain - one resembled by the genetically-engineered version we tested in cultured microglial cells. These are important findings that provide further support for the critical role of sialic acids in modulating the activity of CD33 in the aging brain. They also suggest that disrupting the interaction between CD33 and sialic acids could represent a powerful therapeutic strategy to inhibit the progression of AD pathology, a point that we also emphasized in our recent study.
Together, these findings suggest that CD33 and its role in microglial cells is clearly emerging as a central player in the pathogenesis of AD (see Figure) and opens new therapeutic avenues for treating and preventing this devastating disease.
References:
Bertram L, Lange C, Mullin K, Parkinson M, Hsiao M, Hogan MF, Schjeide BM, Hooli B, Divito J, Ionita I, Jiang H, Laird N, Moscarillo T, Ohlsen KL, Elliott K, Wang X, Hu-Lince D, Ryder M, Murphy A, Wagner SL, Blacker D, Becker KD, Tanzi RE. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet. 2008 Nov;83(5):623-32. PubMed.
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013 May 22;78(4):631-43. PubMed.
View all comments by Ana GriciucMake a Comment
To make a comment you must login or register.