Light and gravity ensure that tree trunks grow straight up while their roots burrow deep underground. What about neurons, which can project up, down, and anywhere in between? What keeps the axon a pole apart from the dendrites? In this week’s Cell, reports from two independent labs arrive at the conclusion that cellular polarity in neurons is achieved, at least partly, by the differential activation of glycogen synthase kinase 3β (GSK3β). This enzyme has been implicated in Alzheimer disease (AD) because it is one of the kinases that phosphorylates tau, the major constituent of neurofibrillary tangles. GSK3β has also been found to be elevated in AD brain tissue (see Leroy et al., 2002).
The link between tau and neuronal polarity may not be serendipitous. Kozo Kaibuchi and colleagues at Nagoya University, Japan, had previously shown that a protein called CRMP-2 (collapsing response mediator protein-2) is essential for regulating axon growth and that it promotes assembly of microtubules (see also Cole et al., 2004). Tau, of course, is a microtubule binding protein. Now, Kaibuchi, first author Takeshi Yoshimura, and colleagues show that GSK3β phosphorylates not only tau but also CRMP-2.
Previous work had shown that the phosphorylation of threonine 514 (Thr514) on CRMP-2 is crucial for controlling its activity, and that this amino acid lies in a GSK3β consensus phosphorylation site. In the present paper, Yoshimura and colleagues showed that coexpressing CRMP-2 and the GSK3β in COS7 cells causes the mediator protein to be phosphorylated on Thr514. They then wondered how this posttranslational modification might affect axonal growth.
Axonal growth seems a bit of a haphazard affair. In culture, neurons extend tiny growth processes in all directions. After about 12-24 hours, one of these processes rapidly extends to form an axon, and in response, the remaining processes take on the features of dendrites. To see how CRMP-2 might affect this process, the authors used fluorescent antibodies to visually track its location in cultured hippocampal neurons. They found that although total CRMP-2 was elevated in axons sprouting from these neurons, levels of the Thr514 phosphorylated form were 40 percent lower in the vicinity of the growth cone as compared to the shaft. The data suggest that the very tip of the axon contains a pool of unphosphorylated CRMP-2.
Could this spatial regulation of phosphorylation explain the difference between growth of axons vs. dendrites? For one thing, the authors showed that phosphorylation of CRMP-2 inhibits its interaction with tubulin, an essential component of the microtubules that are necessary to elongate an axon. For another, when they mutated Thr514 to remove the phosphorylation site, CRMP-2 became much better at stimulating axon outgrowth than was either the wild-type or a permanently phosphorylated mimic. This, too, points to GSK3β-mediated phosphorylation of CRMP-2 as a crucial factor in preventing axon growth.
Indeed, previous reports have demonstrated that inhibition of GSK3β results in enhanced neurite outgrowth (see Munoz-Montano et al.. 1999), and the authors confirmed this using several inhibitors of the kinase, including lithium chloride (see also ARF recent news story on how lithium may reduce production of amyloid-β by inhibiting GSK3α). When the authors added these inhibitors to cultured hippocampal neurons, they found that the number of neurons with tau-1 positive axons increased by up to twofold. They obtained a similar effect using RNAi to knock down GSK3β, whereas expression of a constitutively active kinase (which would phosphorylate CRMP-2) cut the number of neurons with tau-1 positive axons almost in half.
Next, the authors turned to events upstream of GSK3β. They questioned the role of NT-3 and BDNF, which have been shown to inhibit GSK3β in neurons and enhance axonal elongation and branching. Could these cytokines evoke longer axons because they reduce levels of phosphorylated CRMP-2?
To test this, the investigators added NT-3 to hippocampal neurons. They found that levels of phosphorylated CRMP-2 decreased by almost 40 percent in the growth cone, less in the axonal shafts, and not at all in the cell body, supporting the idea that a spatial gradient of phosphorylated CRMP-2 contributes to cell polarity.
All told, the work supports a scenario whereby inactivation of GSK3β by phosphorylation, possibly in response to growth factor signaling, leads to relatively higher levels of unphosphorylated CRMP-2 at axonal growth cones (see diagram). And indeed, in the second paper, Yi Rao and colleagues at the Shanghai Institute of Biological Sciences and the National Institute of Biological Sciences in Beijing, substantially backed up these findings, and then added some.
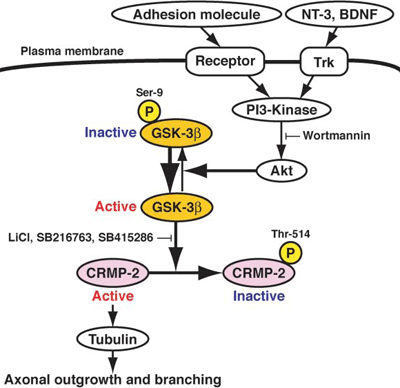
Model for axon growth
Active, unphosphorylated CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. GSK3β can block this by phosphorylating CRMP-2. Signaling events that activate PI3-kinase may stimulate axon growth by activating Akt, which in turn can inactivate GSK3β. [Image courtesy Kozo Kaibuchi.]
Rao’s group found that GSK3β is spatially regulated. In a set of experiments similar to those of the Kaibuchi group on CRMP-2, first author Hui Jiang and colleagues showed that the ratio of inactive GSK3β (phosphorylated at serine 9) to active, unphosphorylated kinase was highest in the tips of the axon. This would explain Kaibuchi’s findings that relatively higher levels of unphosphorylated CRMP-2 (which, remember, is phosphorylated by GSK3β) is driving axon development, and hence, neural polarity.
Moreover, Jiang and colleagues noticed that GSK3β made constitutively active by way of mutation at position 9 led to a decrease in axon formation; half the cultured neurons failed to form axons if they expressed this active kinase, as opposed to only eight percent failure in control cultures. The opposite experiment, inhibiting GSK3β, led to multiple axons. What’s more, these additional axons apparently were functional. Use of the small dye FM4-64 revealed that the axons recycled synaptic vesicles after stimulation with potassium, a sign that they are capable of supporting release of neurotransmitters.
As for upstream signaling events, Jiang found that the kinase Akt also contributes to polarity, again tying in with the findings of Kaibuchi’s group (see, for example, ARF related news story on how Akt may protect neurons in animal models of amyotrophic lateral sclerosis). When Jiang treated cultured hippocampal neurons with LY294002, an inhibitor of phosphatidylinositol-3-kinase (PI3K), he found that not only was phosphorylation of Akt (on serine 473) completely inhibited, but so too was the phosphorylation of GSK3β at serine 9. The authors also found that a constitutively active Akt led to the formation of multiple axons, which would make sense as it would inactivate GSK3β and so lead to more unphosphorylated CRMP-2. As activation of Akt by PI3K can be driven by NT-3/BDNF signaling, this also fits well with the findings of Kaibuchi’s group (see diagram).
While painting a fairly detailed picture of events that impinge on neuronal polarity, these two papers also raise many questions. While “GSK3β activity is of central importance,” as Rao and colleagues write, the complete set of figures remains nebulous for now. What other factors lie downstream of GSK3β—perhaps tau? What about upstream and sidestream? Yoshimura and colleagues showed that GSK3β alone is insufficient for phosphorylation of CRMP-2 in vitro. Other factors are in the mix—perhaps Cdk5, which can phosphorylate CRMP-2 at serines 518 and 522, may be involved? Rao’s group also notes that none of this data explains how Par3/Par6, APC, CDC42 and Rap1B fit in, all of which are known to affect neuronal polarity.
And how might all of this play out in AD or other neurodegenerative diseases? The role of GSK3β in tau hyperphosphorylation is well described, but large gaps remain. “These observations raise the possibility that hyperphosphorylation of CRMP-2 is involved in the development of neurofibrillary tangles and plaque neurites,” suggest Yoshimura and colleagues. This signaling pathway also offers an additional explanation for the neuroprotective effect of BDNF, and suggests that GSK3β inhibitors, in addition to possibly preventing hyperphosphorylation of tau, might also stimulate axonal regeneration.—Tom Fagan
Both Cell papers extend the cascades up- and downstream of GSK-3β considerably by providing molecular links from growth-factor signalling and cell adhesion to microtubule dynamics and axon organization—all most essential processes in evolution in "the making of a brain”!
Deficiency of the GSK-3β ortholog in Drosophila (shaggy/zeste-white 3) causes cells to adopt a neuronal phenotype as "default,” demonstrating the power that GSK-3β kinase has over neuronal development. This influence is further defined by both Cell papers. Most interesting is the appearance of a phosphatase, PTEN, among the wealth of kinases that occupy (litter?) this research field, although with its dual lipid and protein phosphatase activities, PTEN cannot be regarded as a simple addition.
Two more levels of complexity must be recognized. In addition to having its own complex molecular structures—two isozymes in mammals, at least five isoforms of shaggy in Drosophila, and four orthologs of GSK-3 in yeast—GSK-3 activity is regulated by many interacting proteins. On top of this comes active phosphorylation as an extra, dynamic, post-translational control mechanism.
This bewildering intricacy complicates the interpretation of "simple" experiments in mice in vivo, i.e., overexpression or knockout. Complete absence of GSK-3β in mice is lethal for the embryo, probably by suppression of anti-apoptotic actions of NF-κB (Hoeflich et al., 2000). We found that overexpression of constitutively active GSK-3β(S9A) at higher levels was also lethal early in development, while relatively low levels were viable but affected brain size by decreasing the calibre of neuronal soma and apical dendrites (Abstract 42539 Spittaels et al., 2002). We noted no neurodegeneration, however, as opposed to GSK-3β transgenic mice with inducible overexpression to high levels (Lucas et al., 2001). Moreover, GSK-3β could even rescue the axonopathy of tau-4R mice, demonstrating its axonal power in vivo (Spittaels et al., 2000). Combined, the data demonstrate that GSK-3 activity needs to be regulated between narrow boundaries, and that deviation in both directions is dramatic in more than one way and in many organs and multiple systems—a heavy burden on its therapeutic potential.
Although sporadic AD must be regarded as a combination of "accidents" of which the final outcome (amyloid pathology and tauopathy) is due to a multitude of factors, the attraction of the GSK-3 isozymes is their potential to affect both types of pathology, thus providing one trigger… Whether GSK-3α/β isoforms are "accidents" themselves is not clear yet, but likely they are vulnerable partners in the complex signalling cascade outlined by both Cell papers. As part of these cascades, Cdk5 as a priming kinase for GSK-3β does not really explain or help us understand the contributions of either kinase to the tauopathy in AD, but it does reaffirm the link between the two proteins in this respect. We await the final answer: What triggers them to do what they do in aging brain, causing MCI and AD?
1136
In the last issue of Cell, Jiang et al. and Yoshimura et al. have published two excellent articles about the role of GSK3 in determining neuronal polarity.
In most cases, a neuron has a single axon and several dendrites. This type of neuronal polarity is very important for neural network function where signal transmission goes from the axon to the dendrites.
Changes in two cytoskeletal structures, microfilaments and microtubules, have been involved in the establishment of neural polarity. The work of several groups like those of Dotti or Matus has been focused on the role of microfilaments, whereas many other groups have studied the role of microtubules, mainly that of tubulin-binding proteins, in the development of neuronal polarity.
In the first paper, Jiang et al. described the importance of a kinase, GSK3, and a phosphatase, PTEN, in facilitating axonogenesis. In the second paper, Yoshimura et al. have looked for a GSK3 substrate that could be involved in axonogenesis. They found that such a substrate was the tubulin-binding protein CRMP-2, and that CRMP-2 phosphorylation by GSK3 is regulated by trophic factors. In the absence of GSK3 activity, CRMP-2 binds to microtubules, which are stabilized. The consequence of that microtubule stabilization is the cytoplasmic extension that results in the formation of an axon. Thus, the whole axonogenesis mechanism from the ligand (trophic factor) to the morphological change (axonogenesis) could be explained.
These two important observations complement the previous ones on the role of MAP1B, and its modification by GSK3, in axon formation, revealed by several groups like, among others, those of Hirokawa, Edelman, Probst, Salinas, Gordon-Weeks, Fisher, and ourselves. Similar studies on the role of tau protein and also its modification by GSK3 were done by our group and those of Anderton and Bhat, among others.
Also, those observations may suggest the use of GSK3 inhibitors for axonal regeneration.
However, there are yet some questions that remain to be answered, such as why a typical neuron has a single axon and many dendrites.
I think these are very interesting papers from the AD point of view, especially since PS1 is involved in the PI3K/Akt pathway which regulates GSK3 and its downstream target CRMP-2. Specifically it has been shown (see Baki et al., 2004) that PS1 downregulates the activity of GSK3β by stimulating the PI3K/Akt pathway. FAD mutations interfere with the function of PS1 in the PI3K/Akt signaling and this results in upregulation of GSK3β and tau overphosphorylation (Baki et al., 2004).
Since PS1 regulates the PI3K signaling and GSK3 activity, the findings of the Cell papers imply that PS1 may also affect the activity of CRMP-2 (See fig. 7 in Kaibuchi's paper, reproduced above) and this suggests (although it does not prove) that PS1 may also be involved in determining neuronal polarity. Other labs, including that of Dr. Takashima, have also shown that PS1 regulates GSK3 activity.
On the other hand, I find the suggestion of a linkage between CRMP-2 and NFTs a little premature, but reasonable.
Comments
KULeuven
Both Cell papers extend the cascades up- and downstream of GSK-3β considerably by providing molecular links from growth-factor signalling and cell adhesion to microtubule dynamics and axon organization—all most essential processes in evolution in "the making of a brain”!
Deficiency of the GSK-3β ortholog in Drosophila (shaggy/zeste-white 3) causes cells to adopt a neuronal phenotype as "default,” demonstrating the power that GSK-3β kinase has over neuronal development. This influence is further defined by both Cell papers. Most interesting is the appearance of a phosphatase, PTEN, among the wealth of kinases that occupy (litter?) this research field, although with its dual lipid and protein phosphatase activities, PTEN cannot be regarded as a simple addition.
Two more levels of complexity must be recognized. In addition to having its own complex molecular structures—two isozymes in mammals, at least five isoforms of shaggy in Drosophila, and four orthologs of GSK-3 in yeast—GSK-3 activity is regulated by many interacting proteins. On top of this comes active phosphorylation as an extra, dynamic, post-translational control mechanism.
This bewildering intricacy complicates the interpretation of "simple" experiments in mice in vivo, i.e., overexpression or knockout. Complete absence of GSK-3β in mice is lethal for the embryo, probably by suppression of anti-apoptotic actions of NF-κB (Hoeflich et al., 2000). We found that overexpression of constitutively active GSK-3β(S9A) at higher levels was also lethal early in development, while relatively low levels were viable but affected brain size by decreasing the calibre of neuronal soma and apical dendrites (Abstract 42539 Spittaels et al., 2002). We noted no neurodegeneration, however, as opposed to GSK-3β transgenic mice with inducible overexpression to high levels (Lucas et al., 2001). Moreover, GSK-3β could even rescue the axonopathy of tau-4R mice, demonstrating its axonal power in vivo (Spittaels et al., 2000). Combined, the data demonstrate that GSK-3 activity needs to be regulated between narrow boundaries, and that deviation in both directions is dramatic in more than one way and in many organs and multiple systems—a heavy burden on its therapeutic potential.
Although sporadic AD must be regarded as a combination of "accidents" of which the final outcome (amyloid pathology and tauopathy) is due to a multitude of factors, the attraction of the GSK-3 isozymes is their potential to affect both types of pathology, thus providing one trigger… Whether GSK-3α/β isoforms are "accidents" themselves is not clear yet, but likely they are vulnerable partners in the complex signalling cascade outlined by both Cell papers. As part of these cascades, Cdk5 as a priming kinase for GSK-3β does not really explain or help us understand the contributions of either kinase to the tauopathy in AD, but it does reaffirm the link between the two proteins in this respect. We await the final answer: What triggers them to do what they do in aging brain, causing MCI and AD?
References:
Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000 Jul 6;406(6791):86-90. PubMed.
Lucas JJ, Hernández F, Gómez-Ramos P, Morán MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001 Jan 15;20(1-2):27-39. PubMed.
Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, Lasrado R, Vandezande K, Laenen I, Boon T, Van Lint J, Vandenheede J, Moechars D, Loos R, Van Leuven F. Glycogen synthase kinase-3beta phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000 Dec 29;275(52):41340-9. PubMed.
Universidad Autónoma de Madrid
1136
In the last issue of Cell, Jiang et al. and Yoshimura et al. have published two excellent articles about the role of GSK3 in determining neuronal polarity.
In most cases, a neuron has a single axon and several dendrites. This type of neuronal polarity is very important for neural network function where signal transmission goes from the axon to the dendrites.
Changes in two cytoskeletal structures, microfilaments and microtubules, have been involved in the establishment of neural polarity. The work of several groups like those of Dotti or Matus has been focused on the role of microfilaments, whereas many other groups have studied the role of microtubules, mainly that of tubulin-binding proteins, in the development of neuronal polarity.
In the first paper, Jiang et al. described the importance of a kinase, GSK3, and a phosphatase, PTEN, in facilitating axonogenesis. In the second paper, Yoshimura et al. have looked for a GSK3 substrate that could be involved in axonogenesis. They found that such a substrate was the tubulin-binding protein CRMP-2, and that CRMP-2 phosphorylation by GSK3 is regulated by trophic factors. In the absence of GSK3 activity, CRMP-2 binds to microtubules, which are stabilized. The consequence of that microtubule stabilization is the cytoplasmic extension that results in the formation of an axon. Thus, the whole axonogenesis mechanism from the ligand (trophic factor) to the morphological change (axonogenesis) could be explained.
These two important observations complement the previous ones on the role of MAP1B, and its modification by GSK3, in axon formation, revealed by several groups like, among others, those of Hirokawa, Edelman, Probst, Salinas, Gordon-Weeks, Fisher, and ourselves. Similar studies on the role of tau protein and also its modification by GSK3 were done by our group and those of Anderton and Bhat, among others.
Also, those observations may suggest the use of GSK3 inhibitors for axonal regeneration.
However, there are yet some questions that remain to be answered, such as why a typical neuron has a single axon and many dendrites.
Mount Sinai School of Medicine, NYU
I think these are very interesting papers from the AD point of view, especially since PS1 is involved in the PI3K/Akt pathway which regulates GSK3 and its downstream target CRMP-2. Specifically it has been shown (see Baki et al., 2004) that PS1 downregulates the activity of GSK3β by stimulating the PI3K/Akt pathway. FAD mutations interfere with the function of PS1 in the PI3K/Akt signaling and this results in upregulation of GSK3β and tau overphosphorylation (Baki et al., 2004).
Since PS1 regulates the PI3K signaling and GSK3 activity, the findings of the Cell papers imply that PS1 may also affect the activity of CRMP-2 (See fig. 7 in Kaibuchi's paper, reproduced above) and this suggests (although it does not prove) that PS1 may also be involved in determining neuronal polarity. Other labs, including that of Dr. Takashima, have also shown that PS1 regulates GSK3 activity.
On the other hand, I find the suggestion of a linkage between CRMP-2 and NFTs a little premature, but reasonable.
References:
Baki L, Shioi J, Wen P, Shao Z, Schwarzman A, Gama-Sosa M, Neve R, Robakis NK. PS1 activates PI3K thus inhibiting GSK-3 activity and tau overphosphorylation: effects of FAD mutations. EMBO J. 2004 Jul 7;23(13):2586-96. PubMed.
Make a Comment
To make a comment you must login or register.