Whenever a genetic cause is found for a disease, an animal model is in the cards, and Parkinson disease (PD) is no exception. Since recessive mutations in the DJ-1 gene were shown to cause familial PD (see ARF related news story), researchers not only raced to find out what the protein does (see ARF related news story), but also wondered whether knocking it out might prove useful. Wonder no more. In the February 17 Neuron, Jie Shen and colleagues from Brigham and Women’s Hospital, Boston, report the phenotype of DJ-1 knockout mice. The paper is already available online.
Parkinson disease is marked by degeneration and loss of dopaminergic neurons in the substantia nigra of the midbrain, so it is reasonable to expect that DJ-1 knockouts would damage those particular neurons. First author Matt Goldberg and colleagues deleted exon 2 of the DJ-1 gene, which contains the start codon. Yet, when the scientists examined the knockout mice, this part of the brain seemed normal. Even by 12 months of age, the number of dopaminergic neurons had not declined significantly; levels of tyrosine hydroxylase, which catalyzes the rate-limiting step of dopamine synthesis, were also normal. In fact, the amount of dopamine and its metabolites were no different from that found in wild-type mice.
Even so, the animals were not quite right. In the field, they were less active, moving only about 75 percent as much as controls. They were also less likely to get up vertically on their hind legs, rearing about one-third as often as expected. To get at the basis for this behavior, the authors probed dopaminergic neurons with a variety of electrophysiological tests.
First, Goldberg and colleagues looked at dopamine overflow, i.e., the persistence of the neurotransmitter after its release from synapses. They found that it was about sixfold lower in striatal slices from the DJ knockout as compared to control slices. They also found that the dopamine transporter (DAT) blocker nomifensine almost completely restored this overflow, pointing at reuptake of the transmitter. However, they also found that reuptake could not be explained simply by upregulation of the transporter, because both expression of the DAT gene and levels of DAT protein were normal in the DJ knockouts.
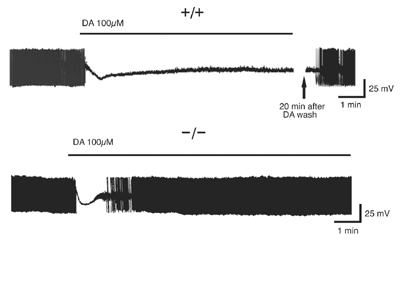
Not all was well with the dopamine receptors, however. When Shen and colleagues incubated nigral neurons with dopamine, it had very different effects on DJ-1-negative than on control neurons. Usually, dopamine activates D2 dopamine autoreceptors. This alters conduction through potassium channels, leading to hyperpolarization of the neuron and blockade of the action potentials (see figure). In DJ-negative neurons, however, this blockade was short-lived and the neurons soon began firing again at full tilt.
D2 receptors fail without DJ-1
When bathed in dopamine, stimulation of D2 dopaminergic autoreceptors normally leads to hyperpolarization of nigral neurons, blocking action potentials and preventing them from firing (top). In DJ-1 knockout neurons, however, the effect of dopamine is short-lived (bottom). The neurons gradually begin to depolarize, and within several minutes they are all firing again, even in the presence of dopamine. [Figure courtesy of Jie Shen and Cell Press]
How might this affect nigral neurons and their connected circuits, such as the glutamatergic medium spiny (MS) neurons in the striatum? When the researchers tested the electrical properties of MS neurons in tissue slices, they found that basal activity was normal in tissue taken from DJ knockouts. But when the same slices were exposed to high-frequency stimulation, the neurons didn’t behave like wild type, which show long-term potentiation (LTP) or long-term depression (LTD), depending on the absence or presence of magnesium. Instead, the DJ-negative neurons only seemed capable of LTP; LTD was absent.
“These results suggest that the LTD impairment is a consequence of reduced D2 receptor-dependent activity, consistent with prior reports that corticostriatal LTD requires both D1 and D2 receptors, whereas corticostriatal LTP induction depends on activation of D1 but not D2 receptors,” write the authors. This seems to fit with recent findings by Paolo Calabresi and colleagues, who reported that in a rat model of PD, those animals that remain in a state of dyskinesia following L-DOPA administration have lost LTD capability (see ARF related news story).
This important link was also highlighted by Emiliana Borrelli, INSERM, France, in a preview to be published in the February 17 Neuron. “Importantly, recent clinical studies based on D2-specific agonist treatments show that, in humans, activation of D2 receptor signaling might be protective to neuronal degeneration in Parkinson’s disease,” Borelli writes (see 2002 Parkinson Study Group).
As for the DJ-knockout as a model for PD, Borrelli adds that none of the loss-of-function mouse models generated to date show degeneration as severe as that seen in PD (see see ARF companion news story and comment below by Mark Cookson and Huaibin Cai). But she adds that it will be exciting to find the mechanism by which DJ-1 impairs D2 receptor-mediated function.—Tom Fagan
Recessive mutations, like those in DJ-1 that are associated with Parkinsonism, are great candidates for making knockout alleles in mice to try and model the disease, as well as understand normal gene function. In this paper, Matt Goldberg, Jie Shen, and their colleagues have produced the first description of a DJ-1 knockout mouse, thus generating a potential model of these rare patients. They show alterations in dopaminergic function in the absence of cell loss in the nigra. This phenotype may be related to the small changes in the dopamine system reported by the same group in parkin mice, but the changes are more dramatic than in the previous model. We are hampered somewhat by the lack of pathology in the patients in being able to really assess how successful this model has been. Clearly this is not a full “PD” phenotype, as there is no TH-positive cell loss, but the fact that there are measurable differences indicates that this might be a workable model if it can be replicated. …More
What needs further clarification is why DJ-1 would have an effect on synaptic events. As several people have pointed out, this protein has such a range of possible functions that it is difficult to immediately see what role it plays at the synapse. It will be nice to see that replacement of the knocked-out DJ-1 rescues the effects, and to investigate whether the known potential for oxidation of the protein is important. There has been some controversy, but it is likely that oxidation of one or more cysteines is important and it would be possible to investigate whether cysteine mutants can perform this normal function of DJ-1.
Another question that might be answered is: How promiscuous is DJ-1 on synaptic function—is it specific for dopamine or does it generally affect all synapses? The data in the paper showing that D2 receptors seem to be preferentially affected hint at specificity, but it would be good to see this confirmed.
Finally, it will be really interesting to see how these mice age and whether there is any effect of crossing them onto other recessive backgrounds (i.e., parkin and PINK1). Do these genes work in tandem to cause additive amounts of disruption, eventually leading to more substantial neuronal loss? Or are they truly a pathway, with each having a sequential effect? Having a model like this gives us perhaps our first opportunity to start to ask some of these questions in a relevant physiological context.
Comments
National Institute on Aging
Recessive mutations, like those in DJ-1 that are associated with Parkinsonism, are great candidates for making knockout alleles in mice to try and model the disease, as well as understand normal gene function. In this paper, Matt Goldberg, Jie Shen, and their colleagues have produced the first description of a DJ-1 knockout mouse, thus generating a potential model of these rare patients. They show alterations in dopaminergic function in the absence of cell loss in the nigra. This phenotype may be related to the small changes in the dopamine system reported by the same group in parkin mice, but the changes are more dramatic than in the previous model. We are hampered somewhat by the lack of pathology in the patients in being able to really assess how successful this model has been. Clearly this is not a full “PD” phenotype, as there is no TH-positive cell loss, but the fact that there are measurable differences indicates that this might be a workable model if it can be replicated. …More
What needs further clarification is why DJ-1 would have an effect on synaptic events. As several people have pointed out, this protein has such a range of possible functions that it is difficult to immediately see what role it plays at the synapse. It will be nice to see that replacement of the knocked-out DJ-1 rescues the effects, and to investigate whether the known potential for oxidation of the protein is important. There has been some controversy, but it is likely that oxidation of one or more cysteines is important and it would be possible to investigate whether cysteine mutants can perform this normal function of DJ-1.
Another question that might be answered is: How promiscuous is DJ-1 on synaptic function—is it specific for dopamine or does it generally affect all synapses? The data in the paper showing that D2 receptors seem to be preferentially affected hint at specificity, but it would be good to see this confirmed.
Finally, it will be really interesting to see how these mice age and whether there is any effect of crossing them onto other recessive backgrounds (i.e., parkin and PINK1). Do these genes work in tandem to cause additive amounts of disruption, eventually leading to more substantial neuronal loss? Or are they truly a pathway, with each having a sequential effect? Having a model like this gives us perhaps our first opportunity to start to ask some of these questions in a relevant physiological context.
Make a Comment
To make a comment you must login or register.