In Optogenetic Mouse, Neural Activity Drives Up Amyloid Deposition
Quick Links
Is too much activity bad for the brain? Stimulated neurons secrete Aβ, and the most metabolically active areas of the brain succumb first to Alzheimer’s pathology. These findings have led to the idea that excess brain activity promotes amyloid deposition, but this has been difficult to demonstrate in vivo. Now, researchers led by Takeshi Iwatsubo at the University of Tokyo have deployed optogenetics to selectively amplify neuronal activity in one side of the brain using flashes of light. As described in the May 12 Cell Reports, five months of repeated stimulation in middle-aged APP transgenic mice more than doubled Aβ deposits in the hippocampus. The findings tighten the link between activity and amyloid. The data also demonstrate that optogenetics, usually used to transiently activate neurons, can be adapted to chronically stimulate a neural pathway in an AD mouse model, Iwatsubo said.
Other researchers praised the approach, saying it holds promise for challenging research questions in vivo. “This is a very exciting use of optogenetics technology, which opens up a new direction for the field to follow,” said Karen Duff, Columbia University, New York. At the same time, she cautioned that, as with any genetic approach, optogenetics studies will require careful controls.
“Like most good studies, it raises more questions than it answers,” said Tara Spires-Jones at the University of Edinburgh. Chief among these are exactly how chronic neuronal activation affects synapses and cognition, and how the data fit with epidemiological studies that correlate more education and lifetime cognitive activity with protection from dementia.
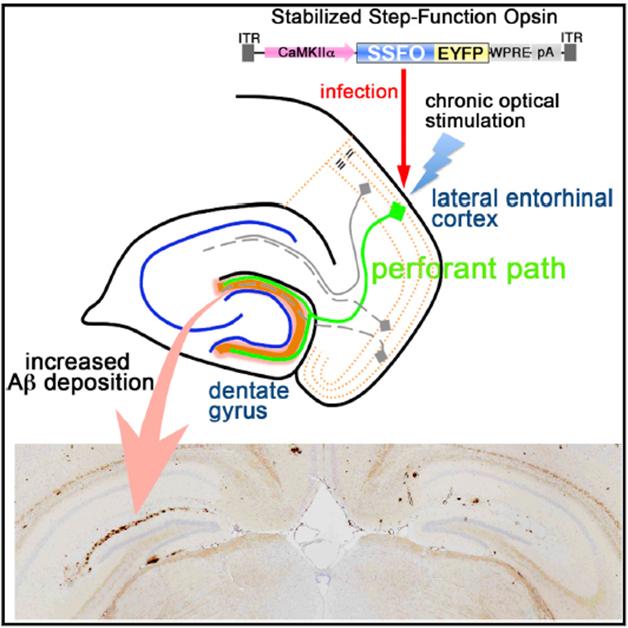
Light Control. Light activation of neurons in the entorhinal cortex stimulates release of Aβ in the dentate gyrus, resulting in more amyloid deposition after several months in the ipsilateral side of the brain (left, lower panel). [Image courtesy of Cell Reports, Yamamoto et al.]
A growing body of research associates amyloid with brain activity. PET imaging studies revealed that deposits appeared first in the interconnected brain regions known as the default mode network (DMN), which are most active when people are not engaged in specific mental tasks (see Feb 2009 news). These regions burn more glucose than other parts of the brain, indicating higher basal activity. Researchers led by David Holtzman and John Cirrito at Washington University, St. Louis, who are co-authors on the current paper, first reported that neuronal activity releases Aβ into interstitial fluid, hinting at a causal connection between amyloid plaques and synaptic activity (see Dec 2005 news). In mouse models, the WashU researchers found that areas of highest spontaneous activity correlated closely with areas where amyloid later accumulated. In addition, suppressing brain activity in sensory regions by trimming a mouse’s whiskers delayed plaque growth in the projection fields of those somatosensory neurons (see May 2011 news). The findings led many to hypothesize a direct connection between activity-dependent Aβ release and amyloid deposition, but it was difficult to obtain evidence for this because researchers lacked ways to selectively and chronically jack up activity in vivo.
Enter optogenetics. This method, developed by co-author Karl Deisseroth at Stanford University in 2005, allows researchers to switch neurons on and off in living mice using light-sensitive ion channels called opsins (see Apr 2007 news; Nov 2012 news series). By expressing the channels in specific neuronal subtypes, researchers gain fine control over which circuits they want to activate. Typically, neuronal activation by opsins lasted for only milliseconds, rendering it less useful for chronic conditions, but in 2011, Deisseroth developed a long-lasting opsin called stabilized step-function opsin. SSFO keeps neurons depolarized and ready to fire for up to 30 minutes after a single light stimulation (see Yizhar et al., 2011).

Long-Distance Communication.
Researchers activate neurons (red) in dentate gyrus of transgenic mice (left panel) by shining light on opsin-expressing afferent neurons (green) in the entorhinal cortex (right panel). [Image courtesy of Cell Reports, Yamamoto et al.]
In collaboration with Deisseroth, Iwatsubo and colleagues introduced either SSFO with the fluorescent label EYFP, or EYFP alone, into the lateral entorhinal cortex of 9½-month-old A7 APP transgenic mice. The AAV viral vector carrying these constructs also contained a CaM kinase II α promoter, limiting expression of the SSFO gene to excitatory neurons. One month after transduction, joint first authors Kaoru Yamamoto and Zen-ichi Tanei inserted a fiber-optic cannula into the entorhinal cortex. To test the circuit, the researchers flashed blue light into the EC for two seconds every minute for four hours.
EC neurons connect to neurons in the dentate gyrus area of the hippocampus, a circuit known as the perforant path. The perforant path represents the main source of cortical input to the hippocampus, and has been studied extensively in Alzheimer’s research in classic experiments dating back to the 1980s. In AD, the circuit degenerates, contributing to memory problems. In response to acute stimulation of this pathway, Aβ42 in hippocampal interstitial fluid spiked up 24 percent for the first hour in animals expressing SSFO, gradually falling back to baseline levels over the next three hours. This demonstrated that the technique worked acutely.
The researchers next wanted to examine the long-term effects of chronic stimulation. They switched on the blue light for two seconds each day in 10½-month-old animals. With SSFO, this brief stimulation amplified neural activity for the next half-hour. After three months, the researchers saw no difference in amyloid deposits between control and SSFO animals. After five months, however, it was another story. Mice with SSFO in their EC had about 2½ times as much amyloid plaque in the dentate gyrus of the stimulated side as the unstimulated side. Control mice, which expressed the EYFP label only but no SSFO, resembled the unstimulated side of SSFO mice.
Levels of the APP cleavage products sAPPβ and sAPPα were unchanged after a single optic stimulation, the authors reported. This suggested that Aβ in the interstitial fluid came from stores in the neuron rather than being newly produced from APP. The authors do not know if the same held true over the length of the five-month experiment. In future work, Iwatsubo will try to determine where Aβ is stored and released, and what cellular mechanisms underlie this.
In separate experiments, the authors transduced 5-month-old transgenic mice with SSFO and stimulated for five months, but in these young animals they saw no amyloid deposits. The results fit with the idea that plaques must be seeded by small Aβ aggregates that only emerge over time and appear late in life in these mice (see Apr 2015 conference news). Other mouse models that aggressively produce Aβ develop plaques earlier.
Many of the mice experienced brief epileptic seizures after light stimulation. Seizures occur in AD patients and mouse models of AD as well, and seem to worsen cognition (see Sep 2007 news; Mar 2015 news). The presence of seizures in this experiment complicates its interpretation, Duff noted. Seizures can damage cells, and injured neurons often release Aβ. Iwatsubo agreed; he is looking for a paradigm that would produce a lower level of stimulation without seizures. Duff suggested that it would be interesting to measure from electrodes in stimulated brain regions to determine the exact amount of activity associated with Aβ release and deposition.
Does chronic stimulation harm cognition? In future studies, Iwatsubo will measure learning and memory in stimulated mice. Since cognitive deficits correlate better with loss of synapses than with the presence of plaques, Iwatsubo will also examine what happens to these structures in the chronically stimulated dentate gyrus. In experiments by other groups, dampening synaptic activity cuts the number of synapses and worsens memory, but it is unclear if chronic activation would also prune synapses, Spires-Jones noted.
Researchers were intrigued by the implications that higher synaptic activity promotes amyloid deposition. Several ongoing clinical studies are testing the efficacy of deep brain stimulation in Alzheimer’s patients, Duff noted. If this stimulation chronically hyperactivates neural circuits, might it do more harm than good? Animal studies examining the effects of DBS on amyloid might help answer this question, Duff suggested.
How can scientists reconcile the finding that high neural activity in these mice worsens pathology with the established finding that education and cognitive stimulation delay Alzheimer’s? The answer is unknown. Some researchers suggest that lifetime cognitive stimulation and experimental neural excitation are two different things. For one, when people engage in specific cognitive tasks, activation of the DMN tends to be suppressed and so might be pathology through this network, Spires-Jones said. Education may also protect in other ways, by amplifying the total number of synapses so the brain can better tolerate some loss, she added.
The findings dovetail with other recent work, noted Marcus Raichle at WashU. He collaborated with Holtzman’s group on a study that found high blood glucose pumped up both neural activity and Aβ production in APPPS1 mice (see Macauley et al., 2015). Type II diabetes, a condition marked by high blood sugar, predisposes to AD (see AlzRisk).
These mouse studies highlight that Aβ secretion is part of normal physiology. Why does it start to precipitate in some people with age? Do high lifetime levels matter most, or the amount of time each day that Aβ is high, Raichle wondered? Studies have shown that extracellular Aβ drops and clearance increases during sleep (e.g., Sep 2009 news; May 2014 news). People sleep less as they age, Raichle pointed out. Is that a risk factor for AD? Iwatsubo is interested in this, as well. “Restoring sleep may be beneficial,” he suggested.
In addition, the type of brain activity might be as important as the amount, Raichle suggested. He noted that the DMN is not just highly active, but also relies heavily on aerobic glycolysis, the incomplete burning of glucose (see Sep 2010 news). The visual cortex is even more metabolically active than the DMN, but uses oxidative phosphorylation to metabolize glucose; it accumulates no plaques. Raichle is currently studying how the metabolism of the DMN changes across the lifespan. “The level and type of activity and where it occurs are fundamental issues in terms of the pathophysiology of this disease,” Raichle said.—Madolyn Bowman Rogers
References
News Citations
- Cortical Hubs Found Capped With Amyloid
- Paper Alert: Synaptic Activity Increases Aβ Release
- Do Overactive Brain Networks Broadcast Alzheimer’s Pathology?
- Flashy Technique Uses Light to Command Neural Firing
- Protein Propagation Real, but Mechanisms Hazy
- Do "Silent" Seizures Cause Network Dysfunction in AD?
- More Evidence That Epilepsy Drug Calms Neurons and Boosts Memory
- Sleep Deprivation Taxes Neurons, Racks Up Brain Aβ?
- Glymphatic Flow, Sleep, microRNA Are Frontiers in Alzheimer’s Research
- Brain Aβ Patterns Linked to Brain Energy Metabolism
Series Citations
Research Models Citations
Paper Citations
- Yizhar O, Fenno LE, Prigge M, Schneider F, Davidson TJ, O'Shea DJ, Sohal VS, Goshen I, Finkelstein J, Paz JT, Stehfest K, Fudim R, Ramakrishnan C, Huguenard JR, Hegemann P, Deisseroth K. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011 Jul 27;477(7363):171-8. PubMed.
- Macauley SL, Stanley M, Caesar EE, Yamada SA, Raichle ME, Perez R, Mahan TE, Sutphen CL, Holtzman DM. Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J Clin Invest. 2015 Jun;125(6):2463-7. Epub 2015 May 4 PubMed.
External Citations
Further Reading
Primary Papers
- Yamamoto K, Tanei Z, Hashimoto T, Wakabayashi T, Okuno H, Naka Y, Yizhar O, Fenno LE, Fukayama M, Bito H, Cirrito JR, Holtzman DM, Deisseroth K, Iwatsubo T. Chronic optogenetic activation augments aβ pathology in a mouse model of Alzheimer disease. Cell Rep. 2015 May 12;11(6):859-65. Epub 2015 Apr 30 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.