Oligodendrocytes Pump Out Aβ42, Worsening Plaques and Synaptotoxicity
Quick Links
Scientists have always presumed that the Aβ in plaques comes solely from neurons. Three recent papers challenge that assumption, identifying another, unexpected source—oligodendrocytes. In the September Nature Neuroscience, scientists led by Klaus-Armin Nave at the Max Planck Institute for Multidisciplinary Sciences, Göttingen, Germany, reported that knocking out the Aβ-producing secretase BACE1 in oligodendrocytes cut the amount of plaque in amyloidosis mice by almost a third. This matches findings by Marc Aurel Busche, University College London, and colleagues in the July 23 PLoS Biology. In their hands, not only did abolishing oligodendrocyte BACE1 cut plaque by a quarter. It also lowered soluble Aβ42 oligomers and rescued neuronal hyperactivity. A third study, from researchers led by Xiangyou Hu and Riqiang Yan at the University of Connecticut Health Center, Farmington, also found a third fewer plaques in mice after deleting BACE1 in oligodendrocytes. That paper is in press at Molecular Neurodegeneration. Yan provided BACE1 mice for Nave’s study and was a co-author on the Nature Neuroscience paper.
- In amyloidosis mice, oligodendrocyte Aβ contributes up to a third of plaque load.
- Soluble Aβ from these cells triggers neuronal hyperactivity.
- Could halting this production delay Alzheimer’s disease?
Two of the groups reported that cultured human oligodendrocytes make as much or more Aβ than do neurons, and that oligodendrocytes in human brain highly express the genes necessary to produce the peptide, suggesting that these cells can beef up plaque production in people as well.
Stephan Lichtenthaler at the German Center for Neurodegenerative Diseases, Munich, called the work outstanding. He suggested exploring the therapeutic potential of oligodendrocyte-specific inhibition of BACE1. “It would be very valuable to test such an approach, which may be a means to avoid the cognitive worsening side effect of high doses of BACE1-targeted inhibitors previously used in clinical trials,” he wrote.

Well-Equipped. In mouse brain, oligodendrocytes (red) make BACE1 (green), an essential enzyme for producing Aβ. Nuclei are blue. [Courtesy of Rajani et al., PLoS Biology.]
Adding to Plaque Load
A few prior papers had suggested that brain cells other than excitatory neurons could promote plaque (Mar 2014 news; Rice et al., 2020). Some homed in on oligodendrocytes, showing these cells secrete Aβ42, but they did not determine precisely what effect this has on pathogenesis (Skaper et al., 2009; Walter et al., 2019; Jul 2023 news). Notably, single-nucleus RNA-Seqnalysis of bipsied human frontal cortex also pointed to amyloidogenic processing of APP in both neurons and oligodendrocytes (Jul 2023 news).
To nail this down, Nave and colleagues generated mice that lacked BACE1 in either oligodendrocytes or excitatory neurons. Joint first authors Andrew Sasmita and Constanze Depp then crossed these animals with the APPNL-G-F model of amyloidosis. At 6 months of age, oligodendrocyte BACE1 knockouts had 30 percent fewer plaques in cortex and hippocampus than did control APPNL-G-F mice. Neuronal BACE1 knockouts, on the other hand, made almost no plaque, despite the continued production of oligodendrocyte Aβ (image below).
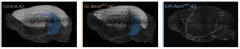
Sharing the Load. Three-dimensional views of the brain show extensive amyloid plaque load in the cortices (white dots) and hippocampi (blue dots) of 6-month-old APPNL-G-F mice (left). They can be cut by a third by knocking out BACE1 in oligodendrocytes (middle), and almost completely by knocking it out in neurons (right). [Courtesy of Sasmita et al., Nature Neuroscience.]
Why no plaques? Previous work suggested that soluble Aβ has to accumulate to a threshold level before plaques form (Lee et al., 2007; Burgold et al., 2014). In the absence of neuronal Aβ, plaque seeding may take longer, Sasmita and colleagues suggested. Supporting this, year-old neuronal BACE1 knockouts did accumulate plaque, but only about a quarter as much as did control APPNL-G-F mice. “[The data] underscore the often-overlooked nonlinear relationship between Aβ production and plaque formation,” Sasmita wrote to Alzforum.
For their part, Busche and colleagues generated similar mice, again crossing the APPNL-G-F line to oligodendrocyte or neuronal BACE1 knockouts. However, first author Rikesh Rajani used a pan-neuronal knockout, rather than one specific to excitatory neurons, and examined plaques in the offspring sooner, at 4 months of age. The findings were almost identical, with oligodendrocyte BACE1 knockouts having a quarter less plaque than controls, and neuronal knockouts having almost none.
In the third study, first author Akihiro Ishii at UConn also crossed APPNL-G-F mice with oligodendrocyte BACE1 knockouts. Ishii used heterozygous APPNL-G-F mice and aged the offspring to one year, but saw the same result, with plaques down about 30 percent in the hippocampus and cortex.
Aβ42 Oligomers, Too
Knocking out oligodendrocyte BACE1 also suppressed soluble Aβ. Nave and colleagues measured about an eighth less Aβ42 in the cortex. Busche and colleagues examined the effects of this soluble peptide by recording synaptic activity in the retrosplenial cortex, an area affected early in AD. To their surprise, oligodendrocyte-specific BACE1 knockout suppressed neuronal hyperactivity, returning neurotransmission to wild-type levels.
Why would oligodendrocytes’ relatively small contribution to soluble Aβ production have such a dramatic effect? Perhaps because, relative to neurons, oligodendrocytes make more Aβ42 than Aβ40. In cultured oligodendrocytes derived from human iPSCs, the Aβ42/Aβ40 ratio was a third higher than in neurons. Since the longer peptide is stickier and more aggregation-prone, oligodendrocyte cultures also generated three times as many soluble Aβ aggregates—oligomers and protofibrils. Injecting this oligodendrocyte-conditioned media into the retrosplenial cortices of wild-type mice triggered hyperactivity.
“The cellular source of Aβ matters,” Busche told Alzforum. “[Our data] challenge the neuron-centric view of AD pathogenesis and underscore the importance of considering oligodendrocytes as active contributors to the disease process,” he said. In future work, he plans to delve into the mechanisms underpinning the higher Aβ42/40 ratio in oligodendrocytes. One possibility is that it has to do with the cells’ membrane lipid composition, which could influence processing of amyloid precursor protein, he speculated.
In addition to making more Aβ42 than did neurons, human oligodendrocytes also made more of the peptide in general, Busche and colleagues found. Induced human oligodendrocytes churned out 50 percent more Aβ than did neurons made from the same iPS lines. Mining publicly available snRNA-Seq data sets, the authors found that oligodendrocytes expressed higher levels of the genes needed to produce Aβ than do neurons. Similarly, when Nave and colleagues analyzed human cortical sections via in situ hybridization, they found that half of oligodendrocytes highly expressed APP and BACE1.
The Next Frontier?
With oligodendrocytes potentially making so much of the Aβ in the human brain, why do they only account for a third of plaque? Andrew Stern at Brigham and Women’s Hospital, Boston, speculated that the local brain environment around cells makes a difference. Oligodendrocytes predominate in white matter, which accumulates few plaques, whereas neurons lie in gray matter, which has many. “This might mean the gray matter interstitial microenvironment is more amyloidogenic than white matter,” Stern wrote to Alzforum (full comment below). If so, modulating this microenvironment could be a therapeutic strategy, he suggested.
For their part, the authors of all three papers said that knockdown of oligodendrocyte-BACE1, specifically, deserves further exploration as a therapeutic strategy. “The next critical step is to show that this approach can actually prevent or rescue disease progression and cognitive decline in mouse models,” Busche wrote. Sasmita agreed with Lichtenthaler that selective knockdown might avoid some of the negative side effects seen in trials with global BACE1 knockdown (Jul 2023 conference news). In mice, deleting the enzyme in oligodendrocytes during development did not hamper myelination or cause other apparent side effects, according to both the Nave and Hu papers.
Others were intrigued by the implications of this research, noting that this cell type has been relatively understudied in AD. “Including oligodendrocytes in Alzheimer’s disease and related dementias research, particularly in ongoing big data efforts, is essential for a comprehensive understanding of these diseases,” Sharyn Rossi and Diane Bovenkamp at BrightFocus Foundation, Clarksburg, Maryland, wrote in a Molecular Neurodegeneration perspective. They noted that recent studies have suggested oligodendrocytes are involved in the earliest stages of the disease, and called for the cells to be incorporated into omics, biomarker, and systems biology studies.—Madolyn Bowman Rogers
References
News Citations
- Aβ: Served Up by More Than Just Excitatory Neurons
- Cortical Biopsies Hint at Start of Alzheimer's 'Cellular Phase'
- Give BACE Inhibitors a Second Chance?
Research Models Citations
Paper Citations
- Rice HC, Marcassa G, Chrysidou I, Horré K, Young-Pearse TL, Müller UC, Saito T, Saido TC, Vassar R, de Wit J, De Strooper B. Contribution of GABAergic interneurons to amyloid-β plaque pathology in an APP knock-in mouse model. Mol Neurodegener. 2020 Jan 8;15(1):3. PubMed.
- Skaper SD, Evans NA, Rosin C, Facci L, Richardson JC. Oligodendrocytes are a novel source of amyloid peptide generation. Neurochem Res. 2009 Dec;34(12):2243-50. PubMed.
- Walter S, Jumpertz T, Hüttenrauch M, Ogorek I, Gerber H, Storck SE, Zampar S, Dimitrov M, Lehmann S, Lepka K, Berndt C, Wiltfang J, Becker-Pauly C, Beher D, Pietrzik CU, Fraering PC, Wirths O, Weggen S. The metalloprotease ADAMTS4 generates N-truncated Aβ4-x species and marks oligodendrocytes as a source of amyloidogenic peptides in Alzheimer's disease. Acta Neuropathol. 2019 Feb;137(2):239-257. Epub 2018 Nov 13 PubMed.
- Lee CC, Nayak A, Sethuraman A, Belfort G, McRae GJ. A three-stage kinetic model of amyloid fibrillation. Biophys J. 2007 May 15;92(10):3448-58. Epub 2007 Feb 26 PubMed.
- Burgold S, Filser S, Dorostkar MM, Schmidt B, Herms J. In vivo imaging reveals sigmoidal growth kinetic of β-amyloid plaques. Acta Neuropathol Commun. 2014 Mar 28;2(1):30. PubMed.
Further Reading
Primary Papers
- Sasmita AO, Depp C, Nazarenko T, Sun T, Siems SB, Ong EC, Nkeh YB, Böhler C, Yu X, Bues B, Evangelista L, Mao S, Morgado B, Wu Z, Ruhwedel T, Subramanian S, Börensen F, Overhoff K, Spieth L, Berghoff SA, Sadleir KR, Vassar R, Eggert S, Goebbels S, Saito T, Saido T, Saher G, Möbius W, Castelo-Branco G, Klafki HW, Wirths O, Wiltfang J, Jäkel S, Yan R, Nave KA. Oligodendrocytes produce amyloid-β and contribute to plaque formation alongside neurons in Alzheimer's disease model mice. Nat Neurosci. 2024 Sep;27(9):1668-1674. Epub 2024 Aug 5 PubMed.
- Rajani RM, Ellingford R, Hellmuth M, Harris SS, Taso OS, Graykowski D, Lam FK, Arber C, Fertan E, Danial JS, Swire M, Lloyd M, Giovannucci TA, Bourdenx M, Klenerman D, Vassar R, Wray S, Sala Frigerio C, Busche MA. Selective suppression of oligodendrocyte-derived amyloid beta rescues neuronal dysfunction in Alzheimer's disease. PLoS Biol. 2024 Jul;22(7):e3002727. Epub 2024 Jul 23 PubMed.
- Ishii A, Pathoulas JA, Omar OMF, Ge Y, Yao AY, Pantalena T, Singh N, Zhou J, He W, Murphy P, Yan R, Hu X. Contribution of amyloid deposition from oligodendrocytes in a mouse model of Alzheimer’s disease. Mol Neurodegener. In press.
- Rossi SL, Bovenkamp DE. Are oligodendrocytes the missing link in Alzheimer’s disease and related dementia research?. Mol Neurodegener. In press.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.