New Data Question Anti-Microbial Powers of Aβ
Quick Links
Is Aβ part of the body’s viral containment system? The idea has drawn plenty of attention, but a paper now reports evidence to the contrary, at least in mice. According to a preprint posted on bioRxiv on January 22, mice with a brain full of Aβ plaques fared no better than their wild-type counterparts when infected with herpes simplex virus-1 (HSV-1). Researchers led by Ilia Baskakov of the University of Maryland School of Medicine in Baltimore reported that in 5xFAD mice, HSV-1 was neither found within Aβ plaques nor did it spur them to form. The findings don’t necessarily negate a connection between viral infections and AD, the authors contend, but they do show that in these specific conditions, Aβ is not an anti-microbial peptide.
- An abundance of Aβ in the brain does not protect against HSV-1 infection.
- In 5xFAD mice, HSV-1 was absent from Aβ plaques and did not incite plaque growth.
- HSV-1 did not replicate in plaque-ridden brain regions.
The idea that microbes, especially herpesviruses, play a part in inciting AD pathogenesis was proposed decades ago, and has gradually picked up steam in the field (Jamieson et al., 1991; Jan 2020 news; Itzhaki et al., 2020). Later studies identified a potential explanation for the link: Sticky Aβ is an anti-microbial peptide, imbued with the capability to contain viral particles (Wozniak et al., 2007; Wozniak et al., 2009; May 2016 news). In the case of herpesviruses, the idea goes, Aβ peptides could theoretically spring into action every time a latent infection reawakens in the brain, ultimately inciting Aβ deposition. In support of this, researchers led by Rudolph Tanzi and the late Robert Moir reported that 5xFAD mice survived longer than did wild-type mice after a lethal dose of HSV-1, and that a sublethal infection spurred Aβ plaques to form prematurely in young mice (Jun 2018 news).
The new paper calls this into question. Baskakov, whose research centers around the biochemical nature of prion propagation, took an interest in the infectious etiology of AD a few years ago. His group’s foray into the topic started with a simple question: Does Aβ protect against viral infection? To address it, first author Olga Bocharova and colleagues took a similar approach to Tanzi and Moir: They intracerebrally infected wild-type and 5xFAD mice with HSV-1. As different viral strains exhibit unique properties, Baskakov used two—17Syn+ and McKrae—which was more lethal. The researchers infected mice with three different doses, ranging from 10-fold below to 10-fold above that predicted to be lethal for 50 percent of the mice (LD50). Regardless of the viral strain, dose, or age of the animals, 5xFAD mice survived no better than wild-type mice. Though not statistically significant, there was a whiff of a survival advantage under one circumstance: when 7- to 10-month-old 5xFAD mice were infected with the highest dose of the McKrae strain, 104 plaque-forming units (PFU), a measure of the number of active viral particles.
In a comment to Alzforum, and Tanzi and William Eimer of Massachusetts General Hospital emphasized this protective trend. In their 2018 study, 5xFAD mice better survived infection with a 10,000-fold higher dose of HSV-1—108 PFU—than did wild-type mice. “Perhaps, if the Bocharova et al. survival studies were expanded to use higher doses of virus, they would also have observed a significant degree of protection against HSV-1 infection by Aβ, as we reported,” Eimer and Tanzi wrote. However, Baskakov noted that such an exceedingly high viral dose—which ultimately killed all of the mice, regardless of genotype—seemed less physiological than the infection conditions used in his study, which hovered around the LD50. Eimer and Tanzi also used a different HSV-1 strain—a fluorescently tagged version of strain F (see de Oliviera et al., 2008, for strain description).
Richard Lathe of the University of Edinburgh agreed that multiple parameters, including viral strains and doses, and the number, sex, and genetic background of mice used in experiments, could underlie discrepancies between the two studies.
Baskakov and colleagues also investigated the idea that Aβ plaques trap viruses. Using antibodies against three different HSV-1 glycoproteins—gB, gD, and gH—the researchers were unable to detect any virus associated with Aβ plaques. One antibody—anti-gB—did appear to detect the virus within plaques; however, subsequent experiments revealed that this antibody also bound to plaques in uninfected 5xFAD mice, suggesting cross-reactivity explained the signal. Furthermore, infecting young 5xFAD mice, which had not yet developed plaques, triggered none. The scientists concluded that, at least in this model, HSV-1 neither associates with Aβ plaques nor spurs their growth.
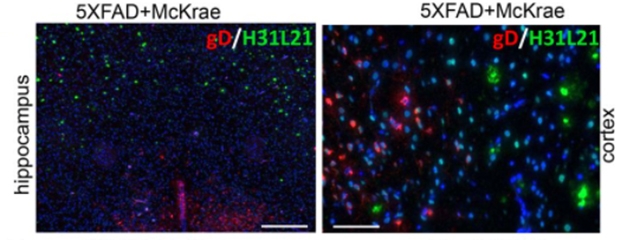
No Overlap. HSV-1 (gD, red) was not found within plaques (green) in the hippocampi of 7- to 10-month-old 5xFAD mice. [Courtesy of Bocharova et al., 2020.]
Interestingly, Baskakov et al. found that, in plaque-ridden 5xFAD mice infected with the highest dose of the McKrae strain—i.e., the condition that hinted at a slight survival advantage—HSV-1 did not appear to replicate within regions of the brain with heavy plaque burden. This was most apparent in the plaque-loaded motor cortex, where the virus was barely detectable in 5xFAD mice despite its abundance there in wild-type mice. The researchers noted that this suppression could explain the smidgen of protection afforded to aged 5xFAD mice.
Tanzi and Eimer believe the virus’s apparent absence from plaques could be explained by methodology. They had visualized it not only with HSV-1 antibodies, but also by using a viral strain expressing a red fluorescent protein (RFP) transgene. They noted that Bocharova et al. did not test whether Aβ’s interaction with HSV-1 glycoproteins may have interfered with binding by the antibodies. Eimer and Tanzi previously reported that Aβ’s anti-viral activities may hinge upon its interactions with HSV-1 surface glycoproteins, implying that Aβ would mask antibody-binding sites on the virus within plaques.
Baskakov emphasized that his findings question Aβ’s anti-viral activity against these two strains of HSV-1 at the doses used. Importantly, acute models of infection don’t reflect the lifelong, latent infections most commonly present in people as they age. His findings leave open the possibility that viruses and other microbes influence the development of AD, via Aβ-dependent or independent mechanisms, he said.
“These studies raise an obvious question: What is the natural target for Aβ antimicrobial action?” asked Lathe. “Antimicrobial proteins differ widely in their activity against different pathogens, and the marginal in vivo protection against HSV-1 in 5xFAD mice (Eimer et al., 2018; and Bocharova et al.) contrasts with the reported strong protective effects against C. albicans and Salmonella,” he added. (Apr 2009 conference news; May 2016 news). “It would certainly be of interest to explore the antimicrobial activity of Aβ against a much wider range of microbes.”—Jessica Shugart
References
News Citations
- Herpes Viruses and Alzheimer’s—The Debate Continues
- Like a Tiny Spider-Man, Aβ May Fight Infection by Cocooning Microbes
- Herpes Triggers Amyloid—Could This Virus Fuel Alzheimer’s?
- Prague: Aβ Rehabilitated as an Antimicrobial Protein?
Paper Citations
- Jamieson GA, Maitland NJ, Wilcock GK, Craske J, Itzhaki RF. Latent herpes simplex virus type 1 in normal and Alzheimer's disease brains. J Med Virol. 1991 Apr;33(4):224-7. PubMed.
- Itzhaki RF, Golde TE, Heneka MT, Readhead B. Do infections have a role in the pathogenesis of Alzheimer disease?. Nat Rev Neurol. 2020 Apr;16(4):193-197. Epub 2020 Mar 9 PubMed.
- Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Lett. 2007 Dec 18;429(2-3):95-100. PubMed.
- Wozniak MA, Mee AP, Itzhaki RF. Herpes simplex virus type 1 DNA is located within Alzheimer's disease amyloid plaques. J Pathol. 2009 Jan;217(1):131-8. PubMed.
- de Oliveira AP, Glauser DL, Laimbacher AS, Strasser R, Schraner EM, Wild P, Ziegler U, Breakefield XO, Ackermann M, Fraefel C. Live visualization of herpes simplex virus type 1 compartment dynamics. J Virol. 2008 May;82(10):4974-90. Epub 2008 Mar 12 PubMed.
- Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, György B, Breakefield XO, Tanzi RE, Moir RD. Alzheimer's Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron. 2018 Jul 11;99(1):56-63.e3. PubMed.
Further Reading
Papers
- Itzhaki R. Herpes simplex virus type 1, apolipoprotein E and Alzheimer' disease. Herpes. 2004 Jun;11 Suppl 2:77A-82A. PubMed.
Primary Papers
- Bocharova O, Pandit NP, Molesworth K, Fisher A, Mychko O, Makarava N, Baskakov IV. Alzheimer's disease-associated β-amyloid does not protect against herpes simplex virus 1 infection in the mouse brain. J Biol Chem. 2021 Jul;297(1):100845. Epub 2021 May 28 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Edinburgh
Bocharova et al. present an interesting perspective on the role of Aβ as an antimicrobial peptide. Rob Moir and colleagues had reported earlier that Aβ has antimicrobial activity in vitro against bacteria and yeast (Soscia et al., 2010), agglutinates Candida albicans cells in vitro (Kumar et al., 2016), and that transgenic Caenorhabditis elegans nematodes expressing human Aβ1–42 show increased survival following challenge with C. albicans (Kumar et al., 2016). In this same paper, they reported that transgenic 5xFAD mice overexpressing human Aβ were differentially protected against challenge with the bacterium Salmonella typhimurium (P = <0.0001). A fivefold reduction of brain bacterial load was seen in the transgenic mice. In a later paper (Eimer et al., 2018) the same team showed that 5xFAD mice (5–6 weeks, n = 14, all female, heterozygous BL6/SJL background) were protected (P = 0.045) against challenge with HSV-1 (strain unspecified; 108/9 PFU of virus), but protection was partial, and all challenged animals eventually succumbed irrespective of genotype.
Bocharova et al. attempted to repeat these experiments, also using 5xFAD mice (5–6 weeks) on a similar genetic background, and two different strains of HSV-1 (17syn+ and McKrae). Although they state that 5xFAD mice were not differentially protected, mice challenged with 104 PFU of HSV-1 McKrae (the highest dose tested for this virus) did in fact show partial protection, and two of seven transgenic mice survived to 350 h post-infection, whereas all control mice (n = 8) died (P = 0.075). Thus, this trends toward confirming the previous report (Eimer et al., 2018), and would probably have achieved P = <0.05 if they had used more animals.
However, these are tricky experiments: In addition to the need for biological containment of live viruses, transgenic mice are finicky and expensive (hence the low numbers used). Mice are not a natural host for HSV-1, and different mouse strains show very different susceptibilities to HSV-1. BL6 mice are said to be highly resistant whereas SJL mice are sensitive (oral infection with 2 × 105 PFU) (Kastrukoff et al., 2012); the mixed BL6 and SJL genetic background therefore adds uncertainty. Furthermore, Bocharova et al. used “random” male and female mice, whereas Eimer et al. only used females; female mice are reported to be 27-fold more susceptible to HSV-1 than male mice (Geurs et al., 2012). In short, individual mice in the experiment of Bocharova et al. could have had different levels of innate susceptibility, potentially confounding interpretation.
Bocharova et al. also reported no correlation between viral load and genotype, but only investigated cortex, which does not express HSV-1 receptors (Lathe and Haas, 2017), where virions presumably arrived by simple diffusion, not through local replication). They also found no co-localization of HSV-1 with Aβ; this might be expected because Aβ expression in 5xFAD mice is predominantly constitutive, whereas in natural settings it is induced by infection/inflammation. Overall, although there are some potential shortcomings, this is a welcome and conscientious study. Indeed, the authors point out that their work does not refute the viral hypothesis of AD.
But are we looking at the right microbe? These studies raise an obvious question: What is the natural target for Aβ antimicrobial action? Antimicrobial proteins differ widely in their activity against different pathogens, and the marginal in vivo protection against HSV-1 in 5xFAD mice (Eimer et al., 2018; and Bocharova et al.) contrasts with the reported strong protective effects against C. albicans and Salmonella. In vitro, Aβ reduced HSV-1 infectivity by only around 30 percent (Eimer et al., 2018; see also Bourgade et al., 2016), but inactivated >90 percent of Enterococcus faecalis and was even better against Candida albicans (Soscia et al., 2010). This could tie in with reports of diverse microbial infections of the brain (Lathe and St. Clair, 2020). It would certainly be of interest to explore Aβ’s antimicrobial activity against a much wider range of microbes, including the spirochete Treponema pallidum and the protozoan Toxoplasma gondii, both of which have been implicated in brain disease.
References:
Bourgade K, Le Page A, Bocti C, Witkowski JM, Dupuis G, Frost EH, Fülöp T Jr. Protective Effect of Amyloid-β Peptides Against Herpes Simplex Virus-1 Infection in a Neuronal Cell Culture Model. J Alzheimers Dis. 2016;50(4):1227-41. PubMed.
Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, György B, Breakefield XO, Tanzi RE, Moir RD. Alzheimer's Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron. 2018 Jul 11;99(1):56-63.e3. PubMed.
Geurs TL, Hill EB, Lippold DM, French AR. Sex differences in murine susceptibility to systemic viral infections. J Autoimmun. 2012 May;38(2-3):J245-53. Epub 2011 Dec 29 PubMed.
Kastrukoff LF, Lau AS, Thomas EE. The effect of mouse strain on herpes simplex virus type 1 (HSV-1) infection of the central nervous system (CNS). Herpesviridae. 2012 Mar 26;3:4. PubMed.
Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med. 2016 May 25;8(340):340ra72. PubMed.
Lathe R, Haas JG. Distribution of cellular HSV-1 receptor expression in human brain. J Neurovirol. 2017 Jun;23(3):376-384. Epub 2016 Dec 15 PubMed.
Lathe R, St Clair D. From conifers to cognition: Microbes, brain and behavior. Genes Brain Behav. 2020 Nov;19(8):e12680. Epub 2020 Jul 9 PubMed.
Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010 Mar 3;5(3):e9505. PubMed.
Massachusetts General Hospital, Harvard
Massachusetts General Hospital
Bocharova et al. tested 5XFAD mice in an attempt to replicate our results demonstrating a protective role of Aβ against HSV1 (Eimer et al., 2018). Bocharova et al. present data intended to suggest the contrary; however, we do not believe their data constitute sufficient contradictory evidence against Aβ in its role as an antimicrobial peptide, or its protective effects against HSV1.
In both our paper and Bocharova et al., survival experiments of 5- to 6-week-old 5xFAD mice were carried out. In contrast to our study, Bocharova et al. claimed no significant protective effects against HSV1 in 5XFAD versus wild-type mice. Bocharova et al. began to observe trends toward enhanced survival at the highest viral load used, i.e., 1x104 PFU. But then, they did not test higher viral loads to replicate the viral load used in our study, which was 1x108 PFU—10,000 times higher. Perhaps, had the Bocharova et al. survival studies extended to higher doses of virus, they also would also have observed a significant degree of protection against HSV1 infection by Aβ, as we reported.
Bocharova et al.’s results are based on immunofluorescent analysis of both young and old 5XFAD mice infected with HSV1, and their inability to observe HSV1 co-localization with Aβ plaques. A significant difference between our study and Bocharova et al. is how HSV1 was visualized. We presented images with HSV1 labeled with a red-fluorescent protein attached to the VP-26 capsid protein, leaving no doubt about its presence or absence. Bocharova et al. used antibodies targeting surface glycoproteins gD and gH. While in our previous studies we showed that Aβ can bind gD and gH, importantly, Bocharova et al. carried out no controls to test whether Aβ interferes with antibody binding, which our unpublished preliminary data suggests. Bocharova et al. cannot properly justify their conclusion that there is no overlap of HSV1 and Aβ in plaques in the absence of this essential control.
Other data presented by Bocharova et al. actually do provide potential support for Aβ’s protective role as an antimicrobial peptide. Immunofluorescent analysis in both Fig. 4 (of young mice) and Fig. 5 (of old mice) presented reduced or absent HSV1 signal in the high Aβ production areas where the Thy1 promoter is most prominent. This could be interpreted as a protective effect being localized to high amyloid concentration areas in the brain.
Fig. S3 revealed reduced Aβ plaque load in HSV1-challenged mice that survived versus unchallenged 5XFAD littermates. Bocharova et al. interpreted this as HSV-1 clearance without triggering Aβ deposition. We interpret this to be due to Aβ’s antimicrobial binding to HSV1, thereby reducing free Aβ levels. This would subsequently delay the formation of amyloid plaques until sufficient concentration for aggregation was once again achieved. Finally, aged 5XFAD mice presented a trend of reduced HSV1 copy number. While not significant, the trend is evident. Perhaps, if Bocharova et al. had increased the number of mice used, the high variance in their results could have been avoided.
Both the role of Aβ as an antimicrobial peptide and the potential role for microbial pathogens in AD are emerging hypotheses in the field right now. Rigorous analysis of the existing work and continued expansion of data is needed to move the field forward in these new directions. In the absence of any in vitro testing of the antimicrobial properties of Aβ, and, in our opinion, the inconclusive in vivo data in 5XFAD mouse, we do not believe Bocharova et al. provide the necessary evidence to refute our conclusions in Eimer et al., 2018. While we disagree with the overall conclusions in Bocharova et al., we do welcome the continued analysis of our hypothesis.
References:
Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, György B, Breakefield XO, Tanzi RE, Moir RD. Alzheimer's Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron. 2018 Jul 11;99(1):56-63.e3. PubMed.
University of Melbourne
To date, I’m not convinced by any of the experimental or clinical evidence that a conventional virus underlies the pathogenesis of AD. Latent or temperate conventional viral activity may, however, play a role in the relatively common phenocopy of AD, marked by TDP43. This condition, variously known as hippocampal sclerosis, frontotemporal dementia, Pick’s disease, FTD/ALS spectrum, FTLD, LATE (and variants thereof) has many of the hallmarks of a latent herpes virus with its topographic propensity to involve the limbic system, possibly associated with a virus-initiated subclinical auto-inflammatory/auto-immune process. The key may lie in the property of TDP43 localizing/binding to foreign nucleic acids in the cytoplasm.
Is AD an unconventional, slow “virus,” i.e., an autocatalytic prion disease of Aβ amyloid? Maybe, but the evidence is hard to come by. Recent cases of Aβ angiopathy linked to childhood neurosurgery and dura mater grafts are beginning to raise awareness of this possibility. The advent of reliable Aβ biomarkers should help shed some light on this.
Do tau and α-synuclein also share this autocatalytic activity? Convincing quantitative evidence for amplification of inoculated “seeds” remains lacking.
Make a Comment
To make a comment you must login or register.