Human Cell Models Reveal Two Sides to α-Synuclein Inclusions
Quick Links
Not all α-synuclein inclusions are created equal. That’s the upshot from a new stem cell approach to study “inclusionopathies,” such as Parkinson’s and Alzheimer’s diseases. As reported in the September issue of Neuron, scientists led by Vikram Khurana at Brigham and Women’s Hospital in Boston honed a method to rapidly differentiate induced pluripotent stem cells (iPSCs) into neurons that quickly develop inclusions. In the case of α-synuclein, these aggregates are strikingly similar to those found in the brains of people who died with a synucleinopathy. They came in two predominant forms: a lipid-rich, unstable form that predominantly haunted neuronal cell bodies, and a stable form that was chock-full of fibrillar α-synuclein aggregates and contained the autophagy protein p62. While the shifty, greasy inclusions were highly toxic to the neurons, the more stable, fibrillar aggregates appeared to be neuroprotective, in some cases merging with the more toxic aggregates and dampening their effect. The authors interpret the findings as evidence that α-synuclein aggregates not only differ in their composition, but also in their effects on the cells that carry them.
- In new models of synucleinopathy, stem cells rapidly form neurons loaded with inclusions.
- Some of these are toxic, some are protective.
- The latter only form when seeded with preformed fibrils.
Khurana and colleagues have used their approach to develop more than 60 inclusionopathy cell models so far, including lines that express different variants of α-synuclein, tau, and other proteins prone to aggregation. Other researchers can use the cells or, even better, use the vectors developed by Khurana to make their own neurons, astrocytes, and other cell types in their own labs, he told Alzforum.
Mark Cookson of the National Institutes of Health in Bethesda, Maryland called the study “state of the art,” and was impressed that the protocols yielded inclusions that resembled those in the brain. That these inclusions have distinct physical properties and toxicities intrigued Cookson, and he thinks they could point the way to therapeutics that take aim at only the harmful aggregates. Cookson co-leads the NIH’s iPSC Neurodegenerative Disease Initiative, which generates a variety of isogenic iPSC lines to model disease risk variants (May 2021 news). He sees Khurana’s approach as complementary to iNDI, which is starting to deploy similar approaches to rapidly create iPSC-derived cells.
Lary Walker of Emory University in Atlanta was also struck by the differential toxicity of the inclusions spotted within the cells. “In light of the expanding technical capacity to generate cell types in vitro that reflect as closely as possible their counterparts in vivo, I have little doubt that these models will play an increasingly important role in mechanistic investigations of neurodegenerative diseases,” he wrote (full comment below).
Khurana thinks iPSC-based cellular models are a powerful way to study disease mechanisms in human cells. However, as the iPSCs are coaxed into neurons, astrocytes, or other desired cell types, they differentiate into heterogenous populations that can vary from dish to dish and from lab to lab. What’s more, the resulting cells are often immature, and often express scant levels of the proteins of interest, including α-synuclein, Khurana said.
To get around some of these problems, co-first authors Isabel Lam and Alain Ndayisaba and colleagues turned to high-capacity piggyBac vectors. These transposable elements can be loaded up with genes, and then, with a dash of transposase, can be integrated into the genome along with their cargo. Using this system, the researchers integrated both NGN2—a transcription factor that switches iPSCs into neurons—and a disease-related gene of choice into the iPSC genome. Treatment with doxycycline then turned them on, simultaneously converting the iPSCs into neurons, and churning out aggregate-prone protein. This approach does not make the neurons more mature relative to neurons induced by other models, Khurana acknowledged. However, they differentiate quicker and expression of the piggyBac genes is robust, he said. For this study, the authors focused on α-synuclein.
Lam and colleagues generated piggyBac constructs loaded with wild-type or mutated forms of α-synuclein, and transfected them into iPSC lines. The paper includes a plethora of findings. What follows are a few of the highlights.
What’s Your Type?
The scientists first compared levels of α-synuclein expressed by NGN2-induced neurons derived from a person who carried an α-synuclein triplication, to levels of wild-type α-synuclein driven by the piggyBac vector. They found that the latter were much higher and closer to levels in cells in postmortem brain samples from people who died with synucleinopathies. To accelerate aggregation in the induced neurons, Lam treated them with preformed α-synuclein fibrils. PFFs induced more inclusions in the neurons with the transgene than it did in neurons with the triplication (image below).
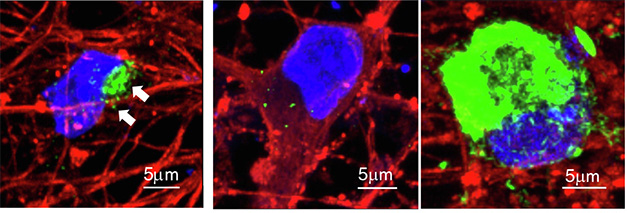
Induced Inclusions. Upon treatment with preformed fibrils, inclusions of phosphorylated α-synuclein (green) formed in iPSC-derived neurons from a patient with an α-synuclein triplication (left), and even more so when α-synuclein was expressed via piggyBac instead (right). No inclusions formed when neurons did not express α-synuclein (middle). [Courtesy of Lam et al., Neuron, 2024.]
In neurons expressing a piggyBac A53T-α-synuclein transgene the scientists found two different types of aggregate (image below). Type I inclusions contained a lipid-rich mix of vesicular structures and mitochondrial membranes, but no p62. These inclusions predominantly resided within the neuronal cell body, and were dynamic, unstable aggregates that dissolved within minutes of exposure to trifluoperazine, an activator of autophagy, or nortriptyline, which prevents α-synuclein aggregation. Both compounds reportedly counter α-synuclein aggregation and toxicity (Höllerhage et al., 2014; Collier et al., 2017). Type II inclusions, on the other hand, were loaded with filamentous α-synuclein but no lipids, and they did not readily dissipate. Some of these, dubbed Type IIa, contained p62 while others, labeled Type IIb, did not. Still other inclusions haunted neurites, and these resembled Type II inclusions in that they were devoid of lipids. Importantly, the researchers found similar types of inclusions in postmortem brain samples from people who died with synucleinopathies.
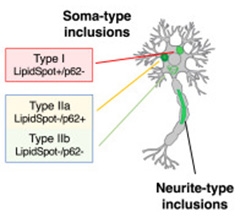
Variety Pack. In the neuronal soma, Type I inclusions were lipid-rich (LipidSpot+) and p62-negative. Type IIa/IIb were lipid-negative. Type IIa harbored p62. Neurites host mostly lipid-negative inclusions. [Courtesy of Lam et al., Neuron, 2024.]
How do these different inclusions interact with each other, and how do they affect cells? Type I, but not type II, inclusions formed in the A53T-α-syn neurons without the addition of PFFs. In some instances, Type I inclusions joined up with the Type II. By tracking both the inclusions and the survival of the cultured neurons over time, the researchers concluded that both Type I and Type IIb inclusions were neurotoxic, while Type IIa, which contains p62, was neuroprotective. The mechanisms underlying this differential toxicity remain a mystery. However, that p62 appears to bestow protective properties hints that α-synuclein degradation by autophagy is beneficial, Khurana suggested.
What controls the formation of these different types of inclusions? And would it be possible to tip the balance from the toxic to protective? These are some of the burning questions the scientists want to answer next. To that end, they are tracing interactions among proteins sequestered within the inclusions, and screening for genetic variants that influence inclusion toxicity. So far, for example, they have tied neurotoxicity to the sequestration of actin cytoskeleton-modulator proteins, such as RhoA, by inclusions.
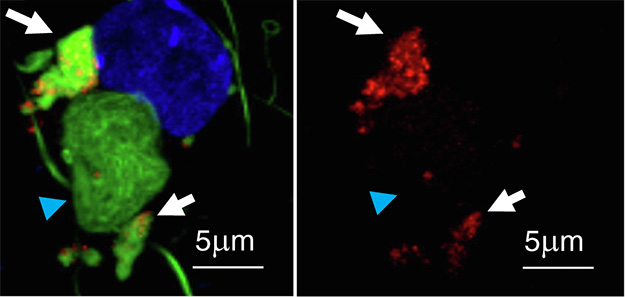
Two Types. Confocal microscopy reveals α-synuclein (green) and lipids (red) in three adjacent inclusions (left and right). The white arrows point to Type I inclusions loaded with lipids. Blue arrowheads indicate a lipid-negative, Type II inclusion. [Courtesy of Lam et al., Neuron, 2024.]
Khurana told Alzforum that the concept of differentially harmful types of α-synuclein inclusions jibes with what scientists have found in human brain samples. Namely, that the pathological burden of α-synuclein doesn’t always track with neurodegeneration. Khurana noted that in people in advanced stages of PD, α-synuclein inclusions riddle the frontal cortex, yet the neurons there are largely spared. Perhaps the types of inclusions that form there tend to be the less toxic, he suggested.—Jessica Shugart
References
News Citations
Paper Citations
- Höllerhage M, Goebel JN, de Andrade A, Hildebrandt T, Dolga A, Culmsee C, Oertel WH, Hengerer B, Höglinger GU. Trifluoperazine rescues human dopaminergic cells from wild-type α-synuclein-induced toxicity. Neurobiol Aging. 2014 Jul;35(7):1700-11. Epub 2014 Jan 28 PubMed.
- Collier TJ, Srivastava KR, Justman C, Grammatopoulous T, Hutter-Paier B, Prokesch M, Havas D, Rochet JC, Liu F, Jock K, de Oliveira P, Stirtz GL, Dettmer U, Sortwell CE, Feany MB, Lansbury P, Lapidus L, Paumier KL. Nortriptyline inhibits aggregation and neurotoxicity of alpha-synuclein by enhancing reconfiguration of the monomeric form. Neurobiol Dis. 2017 Oct;106:191-204. Epub 2017 Jul 12 PubMed.
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Lam I, Ndayisaba A, Lewis AJ, Fu Y, Sagredo GT, Kuzkina A, Zaccagnini L, Celikag M, Sandoe J, Sanz RL, Vahdatshoar A, Martin TD, Morshed N, Ichihashi T, Tripathi A, Ramalingam N, Oettgen-Suazo C, Bartels T, Boussouf M, Schäbinger M, Hallacli E, Jiang X, Verma A, Tea C, Wang Z, Hakozaki H, Yu X, Hyles K, Park C, Wang X, Theunissen TW, Wang H, Jaenisch R, Lindquist S, Stevens B, Stefanova N, Wenning G, van de Berg WD, Luk KC, Sanchez-Pernaute R, Gómez-Esteban JC, Felsky D, Kiyota Y, Sahni N, Yi SS, Chung CY, Stahlberg H, Ferrer I, Schöneberg J, Elledge SJ, Dettmer U, Halliday GM, Bartels T, Khurana V. Rapid iPSC inclusionopathy models shed light on formation, consequence, and molecular subtype of α-synuclein inclusions. Neuron. 2024 Sep 4;112(17):2886-2909.e16. Epub 2024 Jul 29 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.