A Failing Sleep Neuropeptide Hypes Up Hippocampus
Quick Links
As amyloid plaques accumulate in the brain, excitotoxic synaptic activity revs up in the hippocampus. In the June Nature Neuroscience, researchers led by Bart De Strooper and Joris de Wit at KU Leuven, Belgium, identified melanin-concentrating hormone as a stabilizing force that helps counteract this hyperactivity. Produced by the hypothalamus, MCH helps regulate sleep, appetite, and the brain’s reward system.
- The hypothalamic neuropeptide MCH helps suppress hyperactivity in the hippocampus.
- In a mouse model of amyloidosis, this compensatory mechanism fails.
- Amyloid plaques may damage axon terminals, preventing MCH release.
Analyzing their spatial transcriptomics dataset, the authors found an abundance of this cyclic neuropeptide in hypothalamic axon terminals projecting to the hippocampi of young amyloidosis mice. In hippocampal slices, MCH dampened excitatory synaptic transmissions, restoring homeostasis. Later in the disease, this compensatory mechanism failed, and neuronal excitability shot up once more. The work demonstrates how spatial transcriptomics can help parse cellular interactions that may underlie synaptic dysregulation early in disease, noted first author Sara Calafate.
Philip Hasel at New York University agreed. “This is another beautiful example of how sequencing-based spatial transcriptomics, despite its relatively low resolution, can be used to generate, or test, hypotheses,” he wrote (comment below).
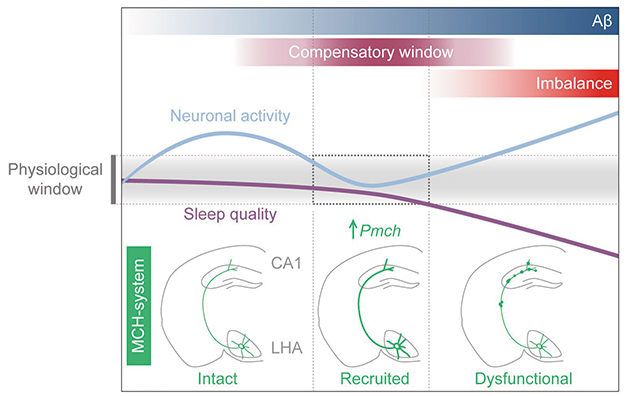
Stabilize For a While. As Aβ deposits in the hippocampus and neuronal activity (blue line) ramps up (left panel), the hypothalamic MCH system and its gene Pmch (green) normalize it (middle). Later in disease (right), the system fails, and hyperactivity and sleep worsen (purple line). [Courtesy of Calafate et al., Nature Neuroscience.]
Previous studies reported that neuronal hyperactivity, sometimes manifesting as epileptic seizures, occurs early in AD (Sep 2007 news; Jul 2013 news; May 2017 news). To study the time course of this phenomenon, Calafate measured activity in hippocampal slices from APPNL-G-F knock-in mice of various ages. Comparing against wild-type tissue, the scientists found higher excitatory activity in slices from 2- and 3-month old knock-ins. Activity came back down to 4 months, then shot back up at 6. This suggested to the authors that compensatory mechanisms manifested between 4 and 6 months (see model above).
To find them, the authors analyzed their spatial transcriptomics dataset from APPNL-G-F mice (Jul 2020 news). At 3.5 months of age, synaptic plasticity genes in their hippocampi were greatly altered. The biggest change was an eightfold boost in the abundance of prepro-melanin concentrating hormone (Pmch) mRNA. This gene makes MCH, and is not expressed in the hippocampus. Instead, the Pmch was in axon terminals projecting from the lateral hypothalamus.
Excitatory hippocampal neurons express the MCH receptor. The authors tested MCH’s effect on them by adding the neuropeptide to hippocampal slices from 3-month-old APPNL-G-F and age-matched wild-type mice. MCH suppressed hyperactivity in APPNL-G-F slices, bringing it back to wild-type levels.
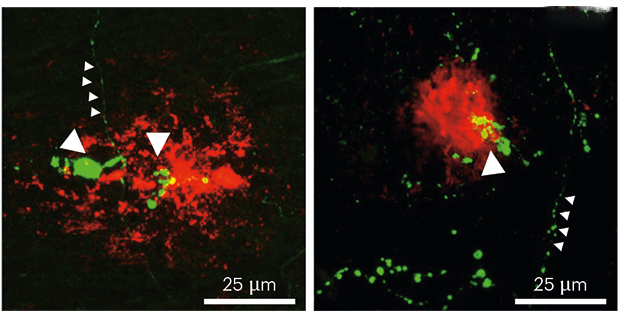
Pernicious Plaques. In mouse (left) and human (right) brain, amyloid plaques (red) cause MCH (green) to accumulate in nerve terminals (large arrowheads). Away from plaques, MCH levels stay low (small arrowheads). [Courtesy of Calafate et al., Nature Neuroscience.]
What changes at 6 months? Examining hypothalamic axon terminals by immunohistochemistry, the authors found that those in the vicinity of amyloid plaques became misshapen, growing blebs filled with MCH. Postmortem AD brain showed similar abnormalities (see image above). Perhaps the spread of amyloid plaques damages axon terminals, preventing the release of MCH, the authors speculated.
The MCH system is not limited to the hippocampus. It projects to several brain regions and regulates multiple behaviors, including sleep. MCH-producing neurons are active during REM sleep, extending its duration. The authors found that 6-month-old APPNL-G-F mice spent less time in REM than did wild-types, again indicating problems with MCH neurons. Calafate noted that this is probably unrelated to the hippocampal function of MCH, because the hippocampus does not influence sleep rhythms.
Sleep problems are part of AD (Aug 2017 conference news; Dec 2017 conference news; Mar 2018 news). Sigrid Veasey at the University of Pennsylvania, Philadelphia, noted that REM sleep drops off in advanced AD, not early in disease as it does in these mice. In other words, different mechanisms may be at work. Veasey suggested studying how MCH may contribute to disease progression in the hippocampus. “A next important step would be to see whether inhibition of MCH neurons, or loss of MCH, alters the temporal progression of hippocampal dysfunction,” she wrote (comment below).—Madolyn Bowman Rogers
References
News Citations
- Do "Silent" Seizures Cause Network Dysfunction in AD?
- Epilepsy in Alzheimer’s Can Be Early and Subtle
- What Lies Beneath: Intracranial Probes Pick Up Hippocampal Seizures in AD
- Paper Alert: Those PIGs! Spatial Transcriptomics Add Human Data
- New Ties between AD and the Stages, Waves, and Molecules of Sleep
- Disturbed Sleep Exerts Toll on Memory and Neurodegeneration
- Does Daytime Drowsiness Foreshadow Aβ Accumulation?
Research Models Citations
Further Reading
News
- In Alzheimer's, More ZZZs Once Wakefulness Neurons Die
- Could Restoring Deep Sleep by Jolting the Brain Ward Off Alzheimer’s?
- Lowering Tau Tips the Brain's Balance of Excitation/Inhibition
- Hippocampal Storms Leave Lasting Marks
- Do Overactive Brain Networks Broadcast Alzheimer’s Pathology?
- Sleep: Too Little, or Too Much, Foreshadows Brain Shrinkage
Primary Papers
- Calafate S, Özturan G, Thrupp N, Vanderlinden J, Santa-Marinha L, Morais-Ribeiro R, Ruggiero A, Bozic I, Rusterholz T, Lorente-Echeverría B, Dias M, Chen WT, Fiers M, Lu A, Vlaeminck I, Creemers E, Craessaerts K, Vandenbempt J, van Boekholdt L, Poovathingal S, Davie K, Thal DR, Wierda K, Oliveira TG, Slutsky I, Adamantidis A, De Strooper B, de Wit J. Early alterations in the MCH system link aberrant neuronal activity and sleep disturbances in a mouse model of Alzheimer's disease. Nat Neurosci. 2023 Jun;26(6):1021-1031. Epub 2023 May 15 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
NYU
This is another beautiful example of how sequencing-based spatial transcriptomics, despite its relatively low resolution, can be used to generate, or test, hypotheses. Here, the platform was used to identify differentially expressed genes in specific anatomical domains. Because their spatial transcriptomics platform is sequencing-based, the authors can unbiasedly identify differentially expressed genes using standard bioinformatic tests on differential gene expression, here edgeR. They use it to show that the pyramidal layer of CA1 is enriched in genes associated with synaptic scaling or homeostatic plasticity in the APPNL-G-F mouse model of Alzheimer’s disease.
This paper is also an outstanding example of how publicly available datasets can be reanalyzed to uncover new biological insights. The sheer amount of data generated in each spatial transcriptomics and single-cell RNA-Seq experiment in laboratories around the world are just waiting for the right question to be asked and the right tools to be applied.
Currently, scientists have to pick between two spatial transcriptomics approaches: either they are sequencing-based, and therefore allow for the unbiased discovery of regionally restricted differentially expressed genes, but don’t offer single-cell resolution (for example 10xGenomics Visium); alternatively, they can use spatial transcriptomics platforms that allow for single-cell resolution but require a set of candidate genes as input, and are therefore restricted to a limited number of known cell, subtype, or substate markers (for example, Vizgen’s MERFISH).
I am looking forward to seeing spatial transcriptomics applied to other diseases that are anatomically restricted or have a focal pathology, such as stroke, spinal cord injury, or stab wound insults. Currently, its most powerful application is in combination with other sequencing modalities, such as single-cell RNA-Seq. Integration across the modalities truly uncovers how regionally-restricted insults affect the local environment at the single-cell level and can be used as a powerful discovery tool in neurobiology.
University of Pennsylvania
The authors are to be congratulated on their overall approach to define mechanisms underlying early abnormalities in Alzheimer's disease by using an amyloid precursor protein (APP) mutant knock-in model and then defining alterations that temporally coincide with impaired responses. Transgenic overexpression in select neuronal populations may miss important changes that occur when the mutations are under the endogenous promoter.
This particular model, the APPKI NL-G-F mouse, has aggressive plaque accumulation at the time when hippocampal hyperactivity was found, which is consistent with the findings of Marc Aurel Busche, showing greater excitability near plaques (Busche et al., 2008).
Here, the authors have not defined a critical role for MCH in Alzheimer's dysfunction/synapse loss, etc. An important step would be to see whether inhibition of MCH neurons, or loss of MCH, alters the temporal progression of hippocampal dysfunction, which may best be observed in a model with slower progression, e.g., in the APP knock-in NL-F model.
Sleep/wake abnormalities that are typically seen early in Alzheimer's disease include insomnia (poor consolidation of nighttime sleep or longer awakenings at night) along with alterations in sleep state-dependent waveforms (slow wave power, spindle amplitude and theta synchrony), and in REM sleep increased postural muscle activity (REM sleep behavior disorder). Loss of REM sleep may be a later finding in advanced Alzheimer's disease.
Sleep abnormalities observed in the present study in the APPKI NL-G-F mice, at an age when amyloid plaques are abundant, did not necessarily emulate sleep findings in mild cognitive impairment or Alzheimer's disease. Rather, differences were limited to reduced REM sleep and impaired homeostatic response to sleep loss. These findings are not consistent with sleep-wake responses to MCH neuro lesioning, which does not alter REM sleep amount but tends to shift more of the total REM sleep to the dark period (normal active/increased wake time for nocturnal mice).
These differences in responses suggest injury to other behavioral state-dependent groups of neurons (cholinergic, noradrenergic, etc., in the APPKI NL-G-F model), which would also be consistent with the work of Lea Grinberg and others showing that many behavioral, state-dependent neuronal groups are injured in Alzheimer's disease (Oh et al., 2022).
It will now be important to go back to the APPKI NL-G-F mouse and define temporal loss of/injury to these other neuromodulatory groups of neurons. A differential sensitivity to injury in this model could also provide clues to the molecular mechanisms of neural injury in Alzheimer's.
References:
Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008 Sep 19;321(5896):1686-9. PubMed.
Oh JY, Walsh CM, Ranasinghe K, Mladinov M, Pereira FL, Petersen C, Falgàs N, Yack L, Lamore T, Nasar R, Lew C, Li S, Metzler T, Coppola Q, Pandher N, Le M, Heuer HW, Heinsen H, Spina S, Seeley WW, Kramer J, Rabinovici GD, Boxer AL, Miller BL, Vossel K, Neylan TC, Grinberg LT. Subcortical Neuronal Correlates of Sleep in Neurodegenerative Diseases. JAMA Neurol. 2022 May 1;79(5):498-508. PubMed.
Make a Comment
To make a comment you must login or register.