Et Tu, Methylene Blue? Drug Only Works as Prophylactic
Quick Links
According to the latest study on tau-transgenic mice, methylene blue, a drug in clinical trials for frontotemporal dementia (FTD) and Alzheimer's disease (AD), stops decline in its tracks—but only when given at the earliest stages. The report in the May 10 Acta Neuropathologica Communications comes from the lab of Eva-Maria and Eckhard Mandelkow at the German Center for Neurodegenerative Diseases (DZNE) in Bonn, Germany. They found that treating their own tau-transgenic mice up to three months before the onset of learning and memory impairments markedly improved their performance compared to untreated controls. However, the drug failed to rescue when given after deficits appeared. The data imply that methylene blue could work only as a preventative treatment in FTD and other tauopathies, including AD.
“It suggests that with methylene blue, you would have to treat early in patients who have Alzheimer’s,” said Eva-Maria Mandelkow, pointing to a need for better biomarkers that catch tau accumulation early. However, this proof-of-concept study also hints that a stronger anti-aggregation compound might treat later in the disease, she said. Alzforum briefly reported on some of these preliminary findings from the International Conference on Alzheimer’s Disease two years ago (see Mar 2013 news story).
Methylene blue is FDA-approved for a number of conditions (for review, see Schirmer et al., 2011). The compound crosses the blood-brain barrier, and reportedly prevents aggregation of a variety of neurodegenerative proteins, including tau, TDP-43, and huntingtin in vitro, and in cell and animal models (Wischik et al., 1996; Arai et al., 2010; Sontag et al., 2012).
It’s not clear how methylene blue works. Some scientists suggest it disrupts tau aggregation by oxidizing it and keeping it in monomer form (Feb 2013 news). Others say it stimulates protein degradation and reduces oxidative damage (Medina et al., 2011; Congdon et al., 2012; Stack et al., 2014). In 2008, Claude Wischik of TauRx Therapeutics in Aberdeen, U.K., presented a Phase 2 trial in which the derivative Rember TM reportedly slowed cognitive decline in people with mild to moderate AD (Aug 2008 news). The modified version LMT-X TM is in Phase 3 for the treatment of behavioral-variant frontotemporal degeneration and AD (Oct 2012 news).
In the current experiment, first authors Katja Hochgräfe and Astrid Sydow wondered how much tau could build up before methylene blue was no longer protective. Would it have to be given long before, shortly before, or even after tau accumulates and symptoms arise? They designed different treatment regimens for two mouse models. The TauΔK mouse expresses a full-length version of human tau, minus lysine 280, that is prone to aggregation, accumulates phosphorylated tau, and develops a learning/memory impairment around 12 months (Eckermann et al., 2007). The TauRDΔK mouse expresses only the ΔK280 repeat domain responsible for tau aggregation. This mouse develops neurofibrillary tangles, its neurons die, and learning and memory problems emerge at around 10 months.
In both models, the tau transgene can be turned off, and the mice learn normally again even if decline had already started (Sydow et al., 2011; Van der Jeugd et al., 2012).
The researchers staggered the time when these mice began getting their daily dose of 20 mg/kg of methylene blue in drinking water. They used six to 11 mice per group, a mix of male and females. The TauΔK mice started as 1½-month-old pups, at 9 months of age (three months before learning problems began), or at 15 months (three months after). The TauRDΔK mice got the treatment starting either at 1½ months of age, or at 15 months, five months after their impairments emerged.
At the end of treatment, when mice were older than 15 months, the researchers compared learning and memory in the Morris water maze to that of untreated transgenic mice and wild-type littermates. They also evaluated general motor and exploration behavior within an open field test. Finally, they analyzed the mouse brains for levels of insoluble, phosphorylated, and misfolded tau, as well as levels of presynaptic and postsynaptic proteins. They also checked whether proteins involved in autophagy, ubiquitin-proteasome function, and oxidative stress rose above normal. All the experiments were carried out in blinded fashion.

Telltale Tau: Phosphorylated tau, stained with the AT180 antibody (brown), fills cell soma of the somatosensory cortex in untreated TauΔK mice (g). It diminishes when methylene blue treatment starts at 1½ months (h), and to a lesser extent, at 9 months (i). [Image courtesy of Hochgräfe et al., 2015.]
Methylene blue only prevented behavioral changes in TauΔK mice if it was given before learning impairments set in. Mice treated beginning at 1½ months moved more actively than untreated transgenics in the open-field test, and better learned and remembered the location of a hidden platform in the Morris water maze. They had less insoluble, misfolded, and phosphorylated tau in the cortex than untreated TauΔK mice (see image above). At the same time, they maintained synaptic protein levels, autophagy rose, and proteasome function went up.
Methylene blue had similar, though smaller, effects in mice that started treatment at 9 months of age. However, in mice that began treatment after behavioral problems had started, the drug did not alter learning or memory, even though levels of phosphorylated, misfolded, and insoluble tau fell. It also failed to restore presynaptic protein levels, and only partially improved protein degradation. In no group did methylene blue boost proteins involved in oxidative stress.
TauRDΔK mice were a different story. None of the methylene blue treatment regimens rescued behavioral deficits, reduced tau levels, or improved protein degradation, even if treatment started when mice were just 1½ months old. To Mandelkow, this means that methylene blue could not keep up with the detrimental effects of the accelerated tau accumulation in these animals. To see if more of the drug helped, the researchers doubled the dose and gave it to the mice at 12 months. Curiously, instead of slowing, the drug increased tau aggregation, implying that too much methylene blue is harmful.
Also surprisingly, wild-type mice given methylene blue learned slightly better in the Morris water maze than their untreated pals, hinting at benefits that go beyond the drug’s anti-aggregation properties. Markers for autophagy and proteasome function rose somewhat, implying that methylene blue could stimulate protein degradation pathways, the authors wrote.
Based on these results, methylene blue may indeed prevent decline in tau-related diseases, Eckhard Mandelkow told Alzforum. However, once tau accumulation reaches a threshold (see image below), cognitive impairment is inevitable, he said. He speculated that this could mean treatment would need to start five years prior to cognitive decline in people.
The lab is screening for better anti-aggregation compounds that could treat ongoing cognitive decline. Because behavior in these mouse strains improves once the transgenes are turned off, suppressing aggregation more robustly could treat later in the disease process. “The results may encourage the development of new compounds which are similar in the mechanism of action, but more effective than methylene blue,” agreed Haruhiko Akiyama, Tokyo Metropolitan Institute of Medical Science (see full comment below).
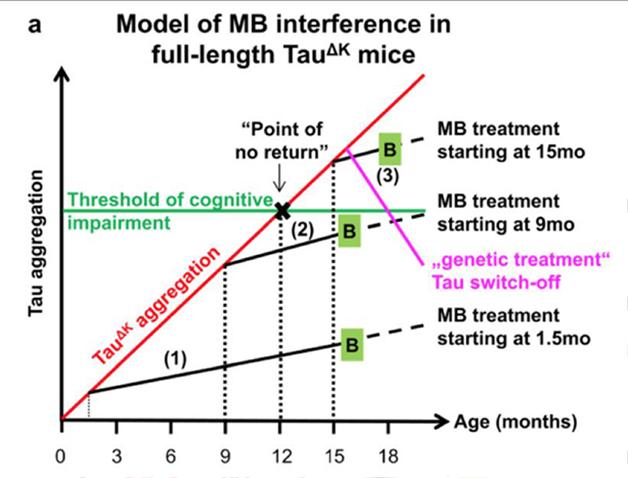
The Model: Tau aggregation reaches a threshold at which cognitive decline starts. Treatment with methylene before, but not after, too much tau builds up prevents decline. In contrast, turning off the toxic tau genes completely seems to reverse symptoms even after they emerge. [Image courtesy of Hochgräfe et al., 2015.]
“This is a very interesting study that sheds some light on the debate about whether methylene blue will be a useful therapeutic for tauopathies,” wrote Tara Spires-Jones, University of Edinburgh, to Alzforum (see full comment below). “It implies that treatment would need to begin very early in the disease course, likely before cognitive impairment is detectable and before substantial pathological changes in tau have occurred.” The LMT-X clinical trials enrolled people with mild to moderate AD, as well as people diagnosed with bvFTD who had frontotemporal atrophy.
“The paper adds to the list of reports now available confirming that treatment with [methylene blue] is able to reverse tau pathology and behavioral deficits in tau transgenic mouse models,” wrote Wischik to Alzforum. He disputes that there is a "point of no return" for treatment, and claims that Hochgräfe and colleagues’ data do not support this because the mice were treated for different lengths of time (only three months in the case of the older mice) and at different ages (see full comment below). The lack of improvement in tau mice after behavioral deficits emerge might suggest that mild to moderate AD patients will fail to respond to LMT-X in ongoing clinical trials. However, Wischik pointed out that the treatments in the oldest mice may not have been given long enough to see any cognitive improvement. His recently published Phase 2 data (Wischik et al., 2015) suggest that patients could benefit even after impairments are evident. He found improvement on the ADAS-Cog in mild and moderate AD patients, and better cerebral blood flow in mild AD patients with six months or more of treatment.
However, catching patients at the right time may not be so easy, Markus Otto of the University of Ulm, Germany, wrote to Alzforum (see full comment below). If the only treatment paradigm for methylene blue is in the early symptomatic or late pre-symptomatic phase, then scientists will need better biomarkers that flag the point of no return, he said. He predicted the ongoing clinical trials of LMT-X will help clarify whether this kind of drug can slow or stabilize cognitive decline in people who already have symptoms.—Gwyneth Dickey Zakaib
References
News Citations
- In Pursuit of Toxic Tau
- Does TauRx Drug Work by Oxidizing Tau?
- Chicago: Out of the Blue—A Tau-based Treatment for AD?
- Will Tau Drug Show Its True Colors in Phase 3 Trials?
Therapeutics Citations
Research Models Citations
Paper Citations
- Schirmer RH, Adler H, Pickhardt M, Mandelkow E. "Lest we forget you--methylene blue...". Neurobiol Aging. 2011 Dec;32(12):2325.e7-16. PubMed.
- Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A. 1996 Oct 1;93(20):11213-8. PubMed.
- Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M, Niizato K, Tsuchiya K, Kobayashi Z, Ikeda K, Yoshida M, Onaya M, Fujishiro H, Akiyama H. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology. 2010 Apr;30(2):170-81. PubMed.
- Sontag EM, Lotz GP, Agrawal N, Tran A, Aron R, Yang G, Necula M, Lau A, Finkbeiner S, Glabe C, Marsh JL, Muchowski PJ, Thompson LM. Methylene blue modulates huntingtin aggregation intermediates and is protective in Huntington's disease models. J Neurosci. 2012 Aug 8;32(32):11109-19. PubMed.
- Medina DX, Caccamo A, Oddo S. Methylene blue reduces aβ levels and rescues early cognitive deficit by increasing proteasome activity. Brain Pathol. 2011 Mar;21(2):140-9. PubMed.
- Congdon EE, Wu JW, Myeku N, Figueroa YH, Herman M, Marinec PS, Gestwicki JE, Dickey CA, Yu WH, Duff KE. Methylthioninium chloride (methylene blue) induces autophagy and attenuates tauopathy in vitro and in vivo. Autophagy. 2012 Apr;8(4):609-22. PubMed.
- Stack C, Jainuddin S, Elipenahli C, Gerges M, Starkova N, Starkov AA, Jové M, Portero-Otin M, Launay N, Pujol A, Kaidery NA, Thomas B, Tampellini D, Beal MF, Dumont M. Methylene blue upregulates Nrf2/ARE genes and prevents tau-related neurotoxicity. Hum Mol Genet. 2014 Jul 15;23(14):3716-32. Epub 2014 Feb 20 PubMed.
- Eckermann K, Mocanu MM, Khlistunova I, Biernat J, Nissen A, Hofmann A, Schönig K, Bujard H, Haemisch A, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem. 2007 Oct 26;282(43):31755-65. Epub 2007 Aug 23 PubMed.
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D'Hooge R, Alzheimer C, Mandelkow EM. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011 Feb 16;31(7):2511-25. PubMed.
- Van der Jeugd A, Hochgräfe K, Ahmed T, Decker JM, Sydow A, Hofmann A, Wu D, Messing L, Balschun D, D'Hooge R, Mandelkow EM. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012 Jun;123(6):787-805. PubMed.
- Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM, Kook KA, Harrington CR. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer's disease. J Alzheimers Dis. 2015;44(2):705-20. PubMed.
External Citations
Further Reading
Papers
- Spires-Jones TL, Friedman T, Pitstick R, Polydoro M, Roe A, Carlson GA, Hyman BT. Methylene blue does not reverse existing neurofibrillary tangle pathology in the rTg4510 mouse model of tauopathy. Neurosci Lett. 2014 Mar 6;562:63-8. Epub 2014 Jan 21 PubMed.
- O'Leary JC, Li Q, Marinec P, Blair LJ, Congdon EE, Johnson AG, Jinwal UK, Koren J, Jones JR, Kraft C, Peters M, Abisambra JF, Duff KE, Weeber EJ, Gestwicki JE, Dickey CA. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. PubMed.
- Cavaliere P, Torrent J, Prigent S, Granata V, Pauwels K, Pastore A, Rezaei H, Zagari A. Binding of methylene blue to a surface cleft inhibits the oligomerization and fibrillization of prion protein. Biochim Biophys Acta. 2013 Jan;1832(1):20-8. Epub 2012 Sep 25 PubMed.
- Jinwal UK, Groshev A, Zhang J, Grover A, Sutariya VB. Preparation and Characterization of Methylene blue Nanoparticles for Alzheimer's Disease and Other Tauopathies. Curr Drug Deliv. 2014;11(4):541-50. PubMed.
Primary Papers
- Hochgräfe K, Sydow A, Matenia D, Cadinu D, Könen S, Petrova O, Pickhardt M, Goll P, Morellini F, Mandelkow E, Mandelkow EM. Preventive methylene blue treatment preserves cognition in mice expressing full-length pro-aggregant human Tau. Acta Neuropathol Commun. 2015 May 10;3:25. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Edinburgh
This is a very interesting study that sheds some light on the debate about whether methylene blue will be a useful therapeutic for tauopathies. Although MB has been reported to reverse tau aggregation in vitro, this study (and others) indicates that while the drug can partially prevent insoluble tau accumulation, it does not remove existing neurofibrillary tangles from the brain. It is hopeful that MB treatment prevented synapse loss and cognitive decline, at least in the less aggressive of the mouse models tested here. In terms of translation, this study implies that treatment would need to begin very early in the disease course, likely before cognitive impairment is detectable and before pathological changes in tau have occurred. This would of course be quite difficult in patients.
View all comments by Tara Spires-JonesChair in Mental Health (Clin.), Aberdeen University
University of Aberdeen
Hochgräfe et al. use tau transgenic mouse models based on constitutive expression of either a full-length 4-repeat tau transgene lacking a lysine at position 280 in the repeat domain (TauΔK) or a repeat domain fragment with the same deletion (TauRDΔK). They showed previously that turning off transgene expression resulted in clearance of tau pathology and resolution of behavioral deficits. Against that background, they sought to determine whether treatment with methylthioninium (MT) as the chloride salt of the oxidized form of MT (MTC) at a nominal dose of 20 mg MTC/kg/day (i.e. ~15 mg MT/kg/day) provided in the drinking water could achieve a similar effect. The rationale is that the aggregation of the exogenous tau in the form of oligomers is responsible for the behavioral deficits, and that MT ought either to prevent aggregation or to enhance clearance of what the authors term pro-aggregant tau.
The results presented succeed in demonstrating this for two of the three treatment durations tested. As such, the paper adds to the list of reports now available confirming that treatment with MT is able to reverse tau pathology and behavioral deficits in tau transgenic mouse models, and their data now support the preventive use of MT treatment.
The authors used three different treatment durations in an attempt to address whether there is a difference between the effectiveness of treatment initiated before or after the appearance of behavioral abnormalities at about 12 months: (1) 14½ months (starting at 1½ months of age), (2) six months (starting at age 9 months) or (3) three months (starting at age 15 months). The effects of treatment are examined at age 16 months for the first two durations, and at 18 months for the shortest treatment duration. Thus, the experimental design does not control for differences due to final age and it can be argued that any difference seen for treatment (3) relative to the other two is due at least in part to more advanced pathology.
Notwithstanding, the authors make a far-reaching claim based on the difference in behavioral effectiveness of treatment (3) versus (1) and (2). They interpret their data as supporting a theory whereby effectiveness of treatment with MT depends critically on whether treatment is initiated before or after behavioral deficits manifest. They claim their data show that whereas treatments (1) and (2) reverse behavioral deficits, treatment (3) does not. Based on this, the authors argue that unlike the “switch-off” paradigm they explored in the earlier report, where time of switch-off made little difference, the effectiveness of MT treatment depends on whether the pathology has reached a “point of no return,” after which treatment is ineffective.
If this were true, it would have major consequences for the potential treatment of AD using MT, in particular for the ongoing Phase 3 clinical trials of a novel form of MT (LMTX®). It would argue that, like the situation that has emerged with the Aβ treatments, clinical efficacy should only be expected for treatment initiated during the preclinical phase of the disease. It would predict that treatment initiated either in the prodromal or the dementia phase would be expected to end up rather like the treatment (3) scenario, with no efficacy due to patients having passed the so-called point of no return.
The authors make this clinical statement in what is otherwise a preclinical paper: “The first anti-Tau therapy with MB in humans was reported by Wischik et al. in 2008 [32]. In this phase 2 clinical trial, a daily dose of 3x 60 mg Rember™ (a derivative of the oxidized form of MB) over 1 year appeared to show a slow-down of cognitive decline in mild and moderate AD patients. Since none of these studies have been published in a peer-reviewed journal, a skeptical attitude towards the presented results remains in the field.”
The data were indeed published, in Journal of Alzheimer’s Disease online on 30 December 2014, and in final form on 21 January 2015 (Wischik et al., 2015). Hochgräfe et al. was submitted on 15 April 2015.
The authors do cite our companion paper (Baddeley et al., 2014), which reports in detail the large body of pharmacokinetic work required to understand the apparent lack of dose-response in the Phase 2 study. This work permitted the discovery that there is, in fact, a simple dose response for the bioavailable dose as opposed to the nominal dose. It was only when this work had been completed that we felt able to publish the Phase 2 data. The Phase 2 efficacy data were reported in part in Baddeley et al., which noted that publication of the full Phase 2 report was imminent. Both papers showed that the minimum effective dose of 138 mg MT/day identified in the Phase 2 study was simply the highest bioavailable dose tested.
Wischik et al. shows that treatment with MT was effective clinically at the primary efficacy endpoint of six months, showing a statistically significant benefit in moderate AD subjects on the ADAS-cog (effect size = -5.42 units, CI = [-9.44, -1.41], nominal p = 0.008, corrected p = 0.047) and on the MMSE (effect size = 3.79 units, CI = [1.16, 6.41], nominal p = 0.0048, corrected p = 0.028). Although there was no evidence of clinical decline in the placebo arm on these scales in mild AD subjects (and therefore no possibility of demonstrating an effect clinically), there was clear evidence of decline in the placebo arm on the functional molecular imaging measure (HMPAO-SPECT) in mild subjects over six months, with a decline in overall signal of -2.16 percent (CI = [−2.72, −1.61]). On this outcome there was a robust treatment effect in mild subjects (effect size = 1.97 percent, CI = [1.02, 2.92], nominal p < 0.001, corrected p < 0.001). At the minimum effective dose, the data provide an empirical estimate of effect size expected in a mild/moderate population at 12 months as approximately 90 percent ± 35 percent (mean ± S.E.) of the expected placebo decline as a basis for planning further studies.
Based on data in humans and pigs (the nearest to humans in terms of pharmacokinetics of MT), the calculated steady-state trough concentration of parent MT and its active metabolites in the human brain at the 138 mg MT/day dose is estimated to be 0.18 μM. This concentration overlaps with the concentrations required for tau aggregation inhibitor activity in vitro as shown by the concentration required for dissolution of PHFs isolated from AD brain tissue (0.16 µM), the Ki of MT for inhibition of aggregation-dependent template-directed proteolysis of tau protein in the physiological milieu of the cell (0.12 µM) in a recently reported cell-based model of tau aggregation (Harrington et al., 2015), and the concentration range over which behavioral deficits are reversed and tau pathology reduced in transgenic tau mouse models that we have reported recently (0.13 – 1.38 µM; Melis et al., 2015).
Thus we respectfully dispute the authors’ claim that treatment with MT is not viable once deficits are manifest. Based on the data summarized here, it appears perfectly viable to achieve reversal of behavioral deficits and pathology in alternative tau transgenic mouse models that cause functional impairment. The concept of pathology being “beyond the point of no return” is therefore not supported by external data.
We would argue that it is also not supported internally by the data presented in Hochgräfe et al. As noted above, the confounding effect of age has not been controlled for in treatment (3). The readout from treatment (3) is undertaken at 18 months, whereas that for the other treatments is at 16 months. Another confounding factor not taken into account is duration of treatment. Treatments (1) and (2) were administered over periods of 14½ and six months respectively, whereas treatment (3) is administered over three months. The obvious conclusion is that for the combination of the TauΔK model, a nominal dose of 20 mg MTC/day, and administration via drinking water, treatment needs to last at least six months. The combination of too short a treatment at a more advanced age would be sufficient to explain the apparent failure of treatment (3).
We believe the behavioral data provided for treatment (3) lack inherent validity. For the exploratory behavior readout, TauΔK without MT and WT without MT are essentially identical in the treatment (3) data. There is therefore no evidence of a phenotype, i.e., both spent 60 percent of the time in the open field. This is different than the data for treatments (1) and (2). As for the water-maze data, a more appropriate analysis than the one presented in this report would be an ANOVA with day and treatment as covariates. Looking at the data for days one through four, there appears to be more of a difference between TauΔK without MT and TauΔK with MT for the supposedly ineffective treatment (3) than for the supposedly effective treatment (2).
Examination of the biochemical data reported for the three treatments suggests there was little difference between different treatment durations on any of the outcomes measured. Thus, there appears to be a reasonably robust effect of MT treatment on both TauΔK and mouse-tau relative to untreated TauΔK as revealed by either antibody K9JA (general tau marker) or TauY9 (human-specific) in the sarkosyl-insoluble fraction. Likewise, there appears to be a generalized strong effect of MT across all treatment durations in terms of reducing levels of phosphorylated mouse tau and phosphorylated TauΔK in the sarkosyl-insoluble brain extract detected by PHF-1. The same absence of differential effect with respect to treatment duration holds true for insoluble TauΔK detected with 12E8 (repeat-domain phosphorylation), although in this case, treatment increases the signal, presumably due to release of tau from oligomers. Similar conclusions hold for the postsynaptic marker PSD95. Insofar as there are effects on autophagy markers, these appear to be most prominent for treatment (2). The only marker for which there appears to be a clear difference between treatment (3) versus treatments (1) and (2) is for the presynaptic protein synaptophysins. Again, this may reflect an effect of treatment duration.
In conclusion, the data reported by Hochgräfe et al. add evidence supporting the view that treatment with MT can reverse behavioral deficits in tau transgenic mouse models. The authors add an elegant, direct demonstration of reduction in insoluble tau (both the human transgene and co-opted endogenous mouse tau) and reversal of its abnormal phosphorylation status. They also add evidence that MT treatment can result in an increase in proteasomal and autophagy markers. Whether this is a direct effect of MT, or a secondary consequence of release of tau from oligomers is not addressed experimentally. The data presented do not support the “point of no return” theory, as any differences between treatments may be a consequence of duration per se.
As regards uncertainty in the field as to the potential utility of treatment approaches based on targeting tau aggregation, this is understandable given the limited data currently available. The critical data will not come from transgenic mice, as the experience with Aβ treatments has demonstrated that there can be efficacy in model systems without translation into clinical efficacy. Even Phase 2 does not resolve the uncertainty, as there have been cases where apparently positive Phase 2 data did not translate into positive Phase 3 results.
We are conducting a Phase 3 program that entails two distinct trials in AD and a trial in behavioral variant frontotemporal dementia, in 1,910 subjects in all. The first of these will report in the first half of 2016. It is only then that we shall learn whether a Phase 3 trial confirms the promising results for this approach seen in Phase 2 and in transgenic mice.
References:
Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM, Kook KA, Harrington CR. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer's disease. J Alzheimers Dis. 2015;44(2):705-20. PubMed.
Baddeley TC, McCaffrey J, Storey JM, Cheung JK, Melis V, Horsley D, Harrington CR, Wischik CM. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer's disease. J Pharmacol Exp Ther. 2015 Jan;352(1):110-8. Epub 2014 Oct 15 PubMed.
Harrington CR, Storey JM, Clunas S, Harrington KA, Horsley D, Ishaq A, Kemp SJ, Larch CP, Marshall C, Nicoll SL, Rickard JE, Simpson M, Sinclair JP, Storey LJ, Wischik CM. Cellular Models of Aggregation-dependent Template-directed Proteolysis to Characterize Tau Aggregation Inhibitors for Treatment of Alzheimer Disease. J Biol Chem. 2015 Apr 24;290(17):10862-75. Epub 2015 Mar 10 PubMed.
Melis V, Magbagbeolu M, Rickard JE, Horsley D, Davidson K, Harrington KA, Goatman K, Goatman EA, Deiana S, Close SP, Zabke C, Stamer K, Dietze S, Schwab K, Storey JM, Harrington CR, Wischik CM, Theuring F, Riedel G. Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav Pharmacol. 2015 Jun;26(4):353-68. PubMed.
View all comments by Charlie HarringtonUniversity of Ulm
Hochgräfe et al. performed an excellent study on timing and dosage of methylene blue in different tau-mutant models. In particular, they developed a disease timing-model (figure 11) for when methylene blue can be an effective drug for cognitive function in mice.
Surprisingly, methylene blue was effective at a time point just before cognitive changes occurred and had only a limited value in long-term treatment. As soon as cognitive changes were present, methylene blue had no or only a limited effect. Very high doses of the drug at a later stage could even induce further aggregation of tau.
What does this mean for treatment options in people? It is difficult to say. One could predict that even in genetic cases, long-term treatment will have no effect; that in symptomatic sporadic patients methylene blue might have no effect; and that the only treatment option would be in the early symptomatic or late presymptomatic phase of sporadic disease. However, how can this be defined? Is a mild cognitive decline already too late, or is that still potentially treatable? We will learn more from the results of the ongoing studies, which include symptomatic patients, and especially from the bvFTD study which included cases with brain atrophy already present.
Certainly we will need better biomarkers to help us define the stage of the disease progress before reaching the point of no return, as defined by the Mandelkows. Therefore, disease-specific follow-up studies are necessary to detect individual changes. For sporadic cases, especially, this will be an enormous effort.
View all comments by Markus OttoWeill Cornell Medical College
We also saw a hormetic dose-response curve with toxicity at high doses. At a dose of 4mg/kg, given before symptoms, we see the same protection in the P301S mice that Hochgräfe et al. showed. We showed improved behavioral abnormalities, tau pathology, inflammation, and oxidative stress. 40mg/kg, identical to the high dose the authors used, was harmful. We and others agree that methylene blue is redox-active at cysteines, and we showed that it activates the nrf2/ARE pathway, which activates anti-oxidant- and reduces inflammation-inducing genes, which was previously unreported. In summary this is a very nicely done study and I agree with their overall conclusions.
View all comments by M. Flint BealTokyo Metropolitan Institute of Medical Science
These results may encourage methylene blue (MB) clinical trials as well as the development of new compounds that have a similar mechanism of action but are more effective than MB. The authors emphasize the importance of administration at early stages, when the mouse strains they employed do not show cognitive impairment. However, Figures 4 and 6 illustrate that all MB groups showed reduction in insoluble and phosphorylated tau. In some cases the difference may not be statistically significant, but the number of mice in each group is small, between three and six. Thus, the results seem to be consistent with our previous study (Hosokawa et al., 2012), in which we started MB administration at the early stage of tau accumulation.
It has to be noted that neurodegenerative lesions consist of a mixture of variable stages of pathology, ranging from normal neurons to those with heavy tau accumulation. In other words, cells at the stages when MB is effective are always present throughout the disease course. It is no wonder then, that if many neurons are already gone, functional deterioration remains even after a disease-modifying therapy is given.
Considering the sensitivity of tau-imaging and other biomarker tests, we will have to start treatment after tau has already accumulated to detectable levels. The present study supports the current therapeutic strategy that targets the preclinical, i.e., symptom-free and biomarker-positive, stage of Alzheimer’s disease.
References:
Hosokawa M, Arai T, Masuda-Suzukake M, Nonaka T, Yamashita M, Akiyama H, Hasegawa M. Methylene blue reduced abnormal tau accumulation in P301L tau transgenic mice. PLoS One. 2012;7(12):e52389. PubMed.
View all comments by Akiyama HaruhitoDZNE, German Center for Neurodegenerative Diseases
It is encouraging that our data obtained in the presented study supports, rather than contradicts, the use of MB as a therapeutic agent for neurodegenerative diseases. In principle our results are in line with most preclinical MB studies published recently. Of course, the interpretation is based on the observations we made using our specific rodent models.
In general, it is important to keep in mind that various Alzheimer’s disease rodent models exist, but none of these models can fully recapitulate a human AD pathology (with plaques and tangles). Especially for sporadic AD cases, which represent the majority of patients and lack pathogenic mutations in APP, PS1 or 2, or Tau, no adequate model organism exists that is suitable for preclinical drug evaluation in terms of cognitive improvements. In this respect, all AD rodent models that exist to date can be considered as somewhat artificial systems. Each recapitulates certain features of the complex pathological spectrum of AD (i.e., by over-expression of pathologic mutations under control of an artificial promoter). Therefore it is problematic to compare the value of different artificial systems and judge whether one or the other is more relevant to the field.
In the case of our mouse models, neither the TauRDΔK mice nor TauΔK animals, expressing the Tau repeat domain or full-length tau with the ΔK280 mutation, respectively, represent a perfect neuropathological correlate to human AD or FTD patients. However, they do reproduce several AD-related neuropathological and functional features quite well, including cognitive dysfunction, synapse loss, as well as Tau missorting, hyperphosphorylation, and neurotoxic accumulation. The higher neurotoxicity is observed in TauRDΔK mice, which can be explained by the high β-propensity due to the pro-aggregant ΔK280 mutation combined with the removal of N- and C-terminal flanking domains.
In both models, the functional impairments could be rescued by "genetic treatment" (i.e., switching off the expression of human Tau) providing proof that Tau pathology can be reversed, in principle, if one removes or neutralizes the toxic Tau species (Sydow et al., 2011; Van der Jeugd et al., 2012). This situation is unique among AD mouse models and offers the potential to study and compare the effects of anti-Tau compounds on Tau aggregation and related functional impairments. The most important finding of our current study was that “preventive” MB treatment can preserve cognition (even if started at a late time point), provided that the tau aggregation propensity is not too strong (see summary of treatment strategies and interpretation in Fig. 11). But beyond these results, the data hold promise that a more potent Tau aggregation inhibitor could rescue cognition even after decline has set in, and perhaps even reach the efficiency of "genetic" treatment.
The term "point of no return" was used in our paper to reflect our experimental observations, but should be taken with a grain of salt, as pointed out in Claude Wischik's commentary. The description as "onset of cognitive decline" would be more neutral. Wischik also emphasizes that the final evaluation of MB and related molecules as potential drugs against neurodegeneration can only be done in humans in the course of clinical studies. Hence, we are looking forward to the results of the various currently ongoing Phase 3 trials, which are announced for late 2015. Wischik and colleagues have developed a variant of MB, called LMTX, which passes more efficiently through the gut and into the brain, and therefore should be superior to MB, as published by the authors in a series of recent papers (e.g. Baddeley et al., 2014; Wischik et al., 2015; and others).
In the meantime, rodent models may provide a “proof-of-concept” and help determine the drug´s mechanism of action. Therefore we included further analysis to figure out underlying beneficial mechanisms, which counteract neurodegeneration. The data point toward a complex interplay of different molecular events that in sum contribute to the preservation of synapses. Especially, MB´s anti-aggregant properties in combination with an increase of protein clearance seem to be of major importance. It is unlikely that the complexity of clinical symptoms observed in AD and other neurological disorders can be fully restored by a single drug. Rather a “cocktail” composed of different compounds, each targeting certain aspects of the disease may lead to successful read-outs.
Finally, we apologize for not citing all of the studies by Wischik and coworkers, which appeared just recently over the last months. Especially, the data of the Phase 2 Rember study presented in Wischik et al.'s JAD report is of major interest for the field. Also the preclinical rodent study published by Melis et al. in Behavioural Pharmacology adds interesting data to the ongoing discussion. In comparison to our data, MB (either applied as MTC or LMTX) was also able to “rescue” behavioral phenotypes of their mouse lines L1 and L66 mice, even after relatively short treatment periods (eight weeks). It is currently not clear which cellular pathways contribute to neuronal regeneration beside MB´s function as tau aggregation inhibitor. Differences between Melis et al. and our studies may be based on the use of different mouse models, which differ in terms of tau constructs and tau mutations used to generate transgenic mice, tau expression levels, tau expression pattern, as well as in certain aspects of tau pathology and behavioral phenotypes. These issues await clarification in future studies.
References:
Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D'Hooge R, Alzheimer C, Mandelkow EM. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011 Feb 16;31(7):2511-25. PubMed.
Van der Jeugd A, Hochgräfe K, Ahmed T, Decker JM, Sydow A, Hofmann A, Wu D, Messing L, Balschun D, D'Hooge R, Mandelkow EM. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012 Jun;123(6):787-805. PubMed.
Baddeley TC, McCaffrey J, Storey JM, Cheung JK, Melis V, Horsley D, Harrington CR, Wischik CM. Complex disposition of methylthioninium redox forms determines efficacy in tau aggregation inhibitor therapy for Alzheimer's disease. J Pharmacol Exp Ther. 2015 Jan;352(1):110-8. Epub 2014 Oct 15 PubMed.
Wischik CM, Staff RT, Wischik DJ, Bentham P, Murray AD, Storey JM, Kook KA, Harrington CR. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer's disease. J Alzheimers Dis. 2015;44(2):705-20. PubMed.
Melis V, Magbagbeolu M, Rickard JE, Horsley D, Davidson K, Harrington KA, Goatman K, Goatman EA, Deiana S, Close SP, Zabke C, Stamer K, Dietze S, Schwab K, Storey JM, Harrington CR, Wischik CM, Theuring F, Riedel G. Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav Pharmacol. 2015 Jun;26(4):353-68. PubMed.
Make a Comment
To make a comment you must login or register.