Do Two APOE4 Alleles Always Mean Alzheimer's?
Quick Links
For 30 years, APOE4 has ranked as the strongest genetic risk factor for Alzheimer’s disease, with two copies boosting the odds up to 15-fold. Now, scientists make the case that people with two APOE4 alleles are not merely at risk, but are destined to develop AD. As reported in Nature Medicine on May 6, researchers led by Juan Fortea and Victor Montal at the Biomedical Research Institute Sant Pau, Barcelona, Spain, compiled neuropathological, biomarker, and clinical evidence from more than 13,000 participants in research studies and clinical trials to assess how APOE4 genotype relates to AD. By age 65, 95 percent of ApoE4 homozygotes had brain amyloid and, on average, memory problems surfaced then, too, a decade sooner than in noncarriers.
- By age 65, 95 percent of APOE4 homozygotes have amyloid plaques.
- This is on par with autosomal-dominant forms of AD.
- On average, symptoms emerge at age 65, a decade earlier than in noncarriers.
- Scientists call APOE4 homozygosity a genetic form of AD.
The penetrance of this age at onset resembles that of autosomal-dominant forms of AD. The genotype is not an absolute dementia predictor because a proportion of APOE4/4 carriers do live into old age without symptoms. Even so, given this data, the authors call for APOE4 homozygosity to be reconceptualized from risk factor to cause. This would elevate APOE4/4 to be the most common genetic cause of AD, affecting up to 3 percent of the world’s population. The study was covered at news outlets across the world.
“Redefining APOE4 homozygosity as a genetic form of Alzheimer’s disease will have a substantial effect on Alzheimer’s disease diagnosis, research, and therapeutic development,” wrote Qin Xu, Zherui Liang, and Yadong Huang of the Gladstone Institute of Neurological Disease in San Francisco, in an editorial in Nature.
Essentially, genetic risk and genetic determination exist on a continuum of sorts. Not everyone is convinced that moving ApoE4/4 from the former to the latter is warranted. For one, while two copies of ApoE4 provoke Alzheimer's pathology with near full penetrance, the extent to which that leads to clinical disease during life is less absolute. For another, most data in the new study came from white participants, but the effect of ApoE4 on AD risk differs by racial and ethnic ancestry. To some scientists, this suggests that ApoE4 homozygosity might not constitute genetically determinant AD. Studies in more diverse populations will tell, they said.
Thirty years from being outed as an AD risk factor, why hasn't this been settled? For one, homozygotes are rare, around 3 percent in the general population and 15 percent among people with Alzheimer’s. Therefore, most AD studies have simply lumped them into one group with heterozygotes. Complicating matters, ApoE4 was categorized a risk factor for late-onset AD. Its effects tend to emerge along with an array of other age-related comorbidities and brain pathologies that contribute to AD risk and cognitive decline. Older onset means many people die of other causes before AD symptoms become obvious, masking the true prevalence in this population. A previous study calculated a 60 percent chance of being diagnosed with AD during life for APOE4 homozygotes (Genin et al., 2011).
Revisiting this question, Fortea and colleagues investigated the penetrance of biological, as opposed to clinical, AD. They collected pathology data from 3,297 brain donors to the National Alzheimer’s Coordinating Center (NACC), 64 percent of whom had had AD dementia. They surveyed cross-sectional fluid and imaging biomarker data from 10,039 people from five clinical cohorts: 2,214 participants in the AD Neuroimaging Initiative (ADNI); 5,477 from the A4 clinical prevention trial; 418 from the ALFA natural history cohort in Barcelona; 612 from the Wisconsin Registry for Alzheimer’s Prevention; and 1,317 from Washington University’s Open Access Series of Imaging Studies (OASIS) 3 Project. Apart from the NACC cohort, 83 percent of participants in the other studies were cognitively unimpaired. For A4, participants were unimpaired at baseline, when these biomarkers were measured. All told, the combined study included 792 ApoE4 homozygotes, more than any other to date.
What did it show? In the NACC sample, 95 percent of APOE4 homozygote but only half the APOE3 homozygote cases had a high or intermediate burden of AD neuropathology (image below). In the clinical cohorts, the researchers looked at fluid and imaging biomarkers, including amyloid-PET, CSF Aβ42, CSF p-tau181. Starting at age 55, APOE4 homozygotes had consistently more abnormal readings than noncarriers; by 65, nearly all APOE4 homozygotes had abnormal CSF Aβ42 and 75 percent had positive amyloid scans. By age 80, 88 percent were positive for all biomarkers. Gene dosage mattered, too, with heterozygotes falling in between noncarriers and homozygotes for both neuropathological and biomarker measures.
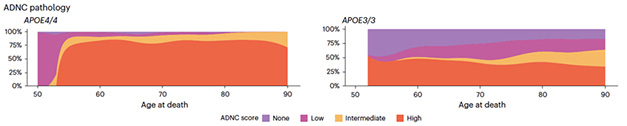
A Penetrating Pathology. In NACC, most APOE4/E4 carriers who died in their mid-50s and beyond had intermediate or high AD neuropathological change (ADNC) scores (left). Half of APOE3/E3 carriers did (right). [Courtesy of Fortea et al., Nature, 2024.]
Next, the scientists asked how ApoE4 influenced the age at which NACC participants and their caregivers reported first noticing symptoms, such as memory problems. On average, first symptoms emerged when homozygotes were 65, followed by MCI at 71, dementia at 73, and death at 77. This pattern started seven to 10 years later in APOE4 noncarriers. Once it did, it proceeded at a similar pace. The finding jibes with numerous studies over the past decades that showed ApoE4 hastens the onset of the AD cascade (for example, Corder et al., 1993; Gomez-Isla et al., 1996; Blacker et al., 1997; Mishra et al., 2018).
How predictable was age at onset? For APOE4 homozygotes, 95 percent developed symptoms within a 32-year span, from age 49 to 81. This seems wide, but is similar to the time span of ADAD mutations, or Down’s syndrome when measured across affected families. Symptoms cropped up over a 47-year span in APOE3 homozygotes.
To monitor the trajectory of biomarker change over the course of disease, the researchers deployed a technique developed for studies of ADAD mutation carriers. They plotted values against estimated years to symptom onset, which they set at 65, the average. The earliest biomarker to shift—CSF Aβ42—was already low in APOE4 homozygotes in their late 40s. These were the youngest people included in the studies, so the researchers were unable to determine when CSF Aβ42 first became abnormal. Next came CSF p-tau181 and amyloid-PET, which turned abnormal in the early 50s.
On MRI, the youngest APOE4 homozygotes in the study had smaller hippocampi than their fellow noncarriers. The scientists ascribe this to neurodevelopmental effects of ApoE4. Later, their hippocampi shrank again, suggesting this atrophy was due to the AD cascade, Fortea told Alzforum. Previous studies have reported smaller regional brain volumes even among infants and young children who carry APOE4 (e.g., Dec 2013 news). Fortea does not know how this developmental influence relates to the atrophy that emerges in adulthood.

Pantheon of Bad. APOE4 homozygosity meets three criteria for genetically determinant forms of Alzheimer's disease, a group that currently includes ADAD and Down’s syndrome. [Courtesy of Xu et al., News and Views, Nature, 2024.]
The data suggests that APOE4 homozygosity meets three criteria for genetic forms of AD: nearly full penetrance, predictability of symptom onset, and predictable sequence of biomarker changes (image above). The authors therefore propose reframing the genetic landscape of AD, counting ApoE4 homozygosity alongside ADAD and Down’s syndrome as determinant forms of the disease. Because one copy of APOE4 causes intermediate disease phenotypes, APOE4 should be considered a semi-dominant gene, they say, as proposed more than a decade ago based on clinical symptoms. This is distinct from autosomal recessive conditions, in which two copies of the disease allele are needed to cause any harm.
Gaël Nicolas and Camille Charbonnier of Normandie University in Rouen, France, believe the findings convincingly demonstrate that APOE4 homozygosity leads to AD biology with near-full penetrance. However, they emphasized that about half of these will not develop dementia by age 85. “That is very important to keep in mind when dealing with such a concept in the clinic, especially with asymptomatic individuals,” they wrote. “In other words, penetrance of AD biology is not penetrance of AD dementia” (comment below).
Fortea and colleagues acknowledge that their study did not assess clinical penetrance. This was not possible in the cohorts at hand, which were biased either toward symptomatic volunteers in the case of NACC, or toward cognitively healthy people in the clinical cohorts. Even so, the near-absolute biological penetrance of APOE4 homozygosity across all studies strengthens the argument that this is a genetically determinant form. AD is increasingly being defined based on biomarkers, not clinical symptoms (Apr 2018 news; Aug 2023 conference news). This biological definition remains controversial, with some taking the view that people who have biomarkers but no symptoms are at risk and not destined to develop dementia (Dubois et al., 2021; Villain and Michalon, 2021; Nov 2023 conference news).
Jessica Langbaum and Eric Reiman of the Banner Alzheimer’s Institute in Phoenix wrote that while Fortea’s findings are consistent with other retrospective case-control studies, prospective cohort studies to date suggest a lower likelihood that APOE4 homozygotes will go on to get MCI or dementia. “For instance, we estimate the lifetime risk (through age 85) of an APOE4 homozygote developing MCI or dementia due to AD at 30 to 55 percent,” they wrote (Qian et al., 2017). They used these numbers to counsel participants in the Alzheimer’s Prevention Initiative’s Generation trial, which specifically enrolled APOE4 carriers (Sep 2016 news). They also noted that at the time of enrollment, a third of APOE4 homozygotes, between the ages of 60 to 75, did not meet the criteria for amyloid positivity. “While more work is needed to understand the differences between cross-sectional and longitudinal study findings, we think it would be inaccurate, and premature, to inform an unimpaired APOE4 homozygote about a nearly certain risk without more longitudinal data in unimpaired persons enrolled prior to age 60 to support this claim,” they wrote.
Langbaum and Reiman acknowledge that APOE4 homozygotes have earlier onset of AD biomarker changes and clinical decline than heterozygotes or noncarriers. They view this as a quantitative, not a categorical, difference from other APOE genotypes. “We don’t find the criteria used to distinguish between the categorical rather than quantitative distinction compelling,” they wrote.
David Holtzman, Washington University in St. Louis, thinks the findings merely further buttress APOE4 as the most powerful genetic risk factor for late-onset AD. Holtzman noted one major difference between APOE4 and autosomal-dominant variants. The risk provoked by the former varies substantially by race and ethnicity, ranging from strongest to weakest in East Asians, non-Hispanic whites, non-Hispanic blacks, and Hispanics (Belloy et al., 2023). In contrast, ADAD mutations, and the trisomy 21 triplication that causes Down’s syndrome, inflict AD equally across ethnoracial groups. “This differential risk … suggests that there must be factors, genetic and other, that can strongly affect risk for different aspects of AD due to APOE4. Given that, I’m not sure, at this point, that it is appropriate to think of APOE4 homozygosity in the same way as autosomal-dominant AD mutations or Down’s syndrome,” he wrote.
Michael Greicius of Stanford University had similar thoughts. “[These ancestral differences] speak to some yet-to-be-discovered genetics and biology that presumably drive this massive difference in risk,” he wrote. “It is unlikely that the authors’ reconceptualization would hold up in an African-ancestry population.” Greicius added that this is important for counseling patients. Fortea and Montal believe more studies are needed to assess how a double dose of APOE4 affects AD risk in diverse groups.
Others question how broadly these findings apply. Ruth Frikke-Schmidt of the University of Copenhagen noted that because the analyses came from AD research cohorts, they may suffer from referral bias, a phenomenon in which people at greatest risk for the disease are more likely to enroll. She called for prospective studies in the general population, which would minimize the risk of overestimating the power of ApoE4.
Colin Masters of the University of Melbourne in Australia said the new study supports the concept—proposed three decades ago—that “sporadic” AD is largely genetic, driven by the APOE4 allele in a dose-dependent fashion (Strittmatter et al., 1993). “In today’s world of Aβ-PET and biofluid biomarkers, we have a clearer understanding that the APOE4 allele drives the onset of Aβ accumulation,” he wrote (comment below).
Fortea and Montal’s proposed reconceptualization would have consequences. The authors contend that because APOE4 homozygotes, regardless of their cognitive status, develop AD pathology in midlife, they should receive disease modifying treatments early and, like ADAD mutation carriers, should be offered enrollment in prevention trials. One fly in the ointment: Lecanemab, the only disease-modifying AD therapy with traditional FDA approval, carries a black-box warning for APOE4 homozygotes due to higher odds of amyloid-related imaging abnormalities (ARIA). Co-author Reisa Sperling of Brigham and Women’s Hospital in Boston, who heads the lecanemab AD prevention trial, AHEAD, said this needs to be addressed. “Even at age 55, APOE4 homozygotes are likely to have AD pathology,” she said. Perhaps treating them early with different dosing regimens, might be able to stave off symptoms while minimizing ARIA, she suggested.
Should APOE4 homozygotes be analyzed in their own right in trials and research studies? “Following this study, APOE4 status must be recognized as a crucial parameter in trial design, patient recruitment and data analysis, with APOE4 homozygotes and heterozygotes being clearly separated,” wrote Huang and colleagues in their editorial. “Such an approach may enhance the treatment efficacy and help tailor therapeutic interventions more effectively toward genetically defined patient populations.”
The ongoing, Phase 3 APOLLOE4 trial of ALZ-801, a follow-on drug of Alzhemed, is being conducted in APOE4 homozygotes, based on hints that the drug may benefit this group specifically. APOE-targeted gene therapy approaches include strategies that aim to reduce ApoE expression, transform APOE4 into the protective APOE2 allele, or overexpress ApoE2 in the hopes of counteracting ApoE4 (Aug 2023 conference news; Dec 2022 conference news; Jackson et al., 2024).
Fortea and Montal do not view ApoE4 homozygosity as a fundamentally different form of AD. Once it starts, the biomarkers and clinical symptoms progress much like in sporadic and familial forms of the disease. That said, Philippe Amouyel of the University of Lille in France thinks it’s possible ApoE4 homozygosity could cause a unique flavor of AD (comment below). “Hence, increased investment in basic research is imperative to fully comprehend how the APOE4 allele, identified over 30 years ago, precisely influences AD pathophysiology,” he wrote. “Once elucidated, powerful new drugs and a straightforward genetic screening test could mitigate the lifetime risk of AD development for APOE4 homozygotes.”—Jessica Shugart
References
Mutations Citations
News Citations
- Brain Volume, Myelination Different in Infants Carrying ApoE4
- New Definition of Alzheimer’s Hinges on Biology, Not Symptoms
- Revised Again: Alzheimer's Diagnostic Criteria Get Another Makeover
- New Alzheimer’s Diagnostic Criteria Remain ‘Research Only’
- To Know or Not to Know: Trial Participants Confront the Question
- Meet the Switching Mice: They Flip Their Glia APOE4 to APOE2
- In Small Trial, Gene Therapy Spurs ApoE2 Production
Therapeutics Citations
Paper Citations
- Genin E, Hannequin D, Wallon D, Sleegers K, Hiltunen M, Combarros O, Bullido MJ, Engelborghs S, De Deyn P, Berr C, Pasquier F, Dubois B, Tognoni G, Fiévet N, Brouwers N, Bettens K, Arosio B, Coto E, Del Zompo M, Mateo I, Epelbaum J, Frank-Garcia A, Helisalmi S, Porcellini E, Pilotto A, Forti P, Ferri R, Scarpini E, Siciliano G, Solfrizzi V, Sorbi S, Spalletta G, Valdivieso F, Vepsäläinen S, Alvarez V, Bosco P, Mancuso M, Panza F, Nacmias B, Bossù P, Hanon O, Piccardi P, Annoni G, Seripa D, Galimberti D, Licastro F, Soininen H, Dartigues JF, Kamboh MI, Van Broeckhoven C, Lambert JC, Amouyel P, Campion D. APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. 2011 Sep;16(9):903-7. Epub 2011 May 10 PubMed.
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993 Aug 13;261(5123):921-3. PubMed.
- Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, Perls TT, Lipsitz LA, Hyman BT. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer's disease. Ann Neurol. 1996 Jan;39(1):62-70. PubMed.
- Blacker D, Haines JL, Rodes L, Terwedow H, Go RC, Harrell LE, Perry RT, Bassett SS, Chase G, Meyers D, Albert MS, Tanzi R. ApoE-4 and age at onset of Alzheimer's disease: the NIMH genetics initiative. Neurology. 1997 Jan;48(1):139-47. PubMed.
- Mishra S, Blazey TM, Holtzman DM, Cruchaga C, Su Y, Morris JC, Benzinger TL, Gordon BA. Longitudinal brain imaging in preclinical Alzheimer disease: impact of APOE ε4 genotype. Brain. 2018 Jun 1;141(6):1828-1839. PubMed.
- Dubois B, Villain N, Frisoni GB, Rabinovici GD, Sabbagh M, Cappa S, Bejanin A, Bombois S, Epelbaum S, Teichmann M, Habert MO, Nordberg A, Blennow K, Galasko D, Stern Y, Rowe CC, Salloway S, Schneider LS, Cummings JL, Feldman HH. Clinical diagnosis of Alzheimer's disease: recommendations of the International Working Group. Lancet Neurol. 2021 Jun;20(6):484-496. Epub 2021 Apr 29 PubMed.
- Villain N, Michalon R. What is Alzheimer's disease? An analysis of nosological perspectives from the 20th and 21st centuries. Eur J Neurol. 2024 Nov;31(11):e16302. Epub 2024 Apr 15 PubMed.
- Qian J, Wolters FJ, Beiser A, Haan M, Ikram MA, Karlawish J, Langbaum JB, Neuhaus JM, Reiman EM, Roberts JS, Seshadri S, Tariot PN, Woods BM, Betensky RA, Blacker D. APOE-related risk of mild cognitive impairment and dementia for prevention trials: An analysis of four cohorts. PLoS Med. 2017 Mar;14(3):e1002254. Epub 2017 Mar 21 PubMed.
- Belloy ME, Andrews SJ, Le Guen Y, Cuccaro M, Farrer LA, Napolioni V, Greicius MD. APOE Genotype and Alzheimer Disease Risk Across Age, Sex, and Population Ancestry. JAMA Neurol. 2023 Dec 1;80(12):1284-1294. PubMed.
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993 Mar 1;90(5):1977-81. PubMed.
- Jackson RJ, Keiser MS, Meltzer JC, Fykstra DP, Dierksmeier SE, Hajizadeh S, Kreuzer J, Morris R, Melloni A, Nakajima T, Tecedor L, Ranum PT, Carrell E, Chen Y, Nishtar MA, Holtzman DM, Haas W, Davidson BL, Hyman BT. APOE2 gene therapy reduces amyloid deposition and improves markers of neuroinflammation and neurodegeneration in a mouse model of Alzheimer disease. Mol Ther. 2024 May 1;32(5):1373-1386. Epub 2024 Mar 19 PubMed.
Further Reading
Papers
- Vance JM, Farrer LA, Huang Y, Cruchaga C, Hyman BT, Pericak-Vance MA, Goate AM, Greicius MD, Griswold AJ, Haines JL, Tcw J, Schellenberg GD, Tsai LH, Herz J, Holtzman DM. Report of the APOE4 National Institute on Aging/Alzheimer Disease Sequencing Project Consortium Working Group: Reducing APOE4 in Carriers is a Therapeutic Goal for Alzheimer's Disease. Ann Neurol. 2024 Apr;95(4):625-634. Epub 2024 Jan 5 PubMed.
Primary Papers
- Fortea J, Pegueroles J, Alcolea D, Belbin O, Dols-Icardo O, Vaqué-Alcázar L, Videla L, Gispert JD, Suárez-Calvet M, Johnson SC, Sperling R, Bejanin A, Lleó A, Montal V. APOE4 homozygozity represents a distinct genetic form of Alzheimer's disease. Nat Med. 2024 May;30(5):1284-1291. Epub 2024 May 6 PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.