Do Motor Neuron Firing Rates Rise, Then Crash, in ALS?
Quick Links
Early on in amyotrophic lateral sclerosis motor, neurons fire like machine guns, but then they run out of ammo and go silent, according to a paper in the January 12 Nature Communications. Researchers from the University of St. Andrews in Scotland saw this pattern in motor neurons expressing TDP-43 mutations or C9ORF72 expansions, two genetic causes of ALS. They tracked these neurons over time in culture, and saw their firing rates initially elevate and then plunge, all before the cells showed other signs of disease. Finding the same problem in both types of neuron suggests that a common pathway could be at work in two kinds of familial ALS. These cultures would be a good testing ground for therapeutics, said study senior author Gareth Miles.
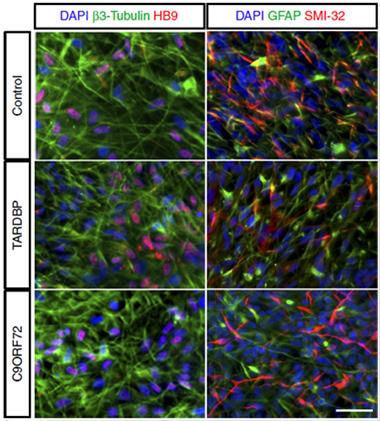
Making Motor Neurons.
The cultures derived from iPS cells of control donors and people with ALS express the proper biomarker profile for motor neurons. [Image courtesy of Nature Communications, Devlin et al.]
Hamstrung by imperfect animal models, scientists have become excited to test theories and treatments in induced pluripotent stem (iPS) cells derived from people with disease. They can give those iPS cells a neural destiny to mimic neurodegenerative diseases in a dish (see Sep 2010 news). However, early experiments on the biochemistry of ALS neurons found they were not very different from control cells, Miles said. More recently, he and others have looked at electrical activity. One group observed diminished excitability in neurons with C9ORF72 expansions, while another reported hyperexcitability in cells with SOD1, C9ORF72, or FUS mutations (Sareen et al., 2013; Wainger et al., 2014). However, because the researchers looked at different time points in the life of the neurons, it is possible the cells may exhibit different activity at different stages.
In the new study, first author Anna-Claire Devlin and colleagues followed cells for 10 weeks. First, collaborators in the laboratory of Siddharthan Chandran at the University of Edinburgh collected skin fibroblasts from one person with ALS due to a TDP-43 mutation, two with C9ORF72-based ALS, and three healthy control donors. They turned the fibroblasts into iPS cells and then pushed them toward a motor neuron lineage. Then Devlin took over, spreading the cells on plates to allow the final steps toward a motor neuron fate (see image above).
After a couple of weeks, many cells matured to a point where they were electrically active, firing repeated action potentials in response to stimulation. However, the ALS lines shot off more frequent action potentials for a given stimulation than the control lines. “They are definitely more excitable,” Miles concluded.
However, as the cells aged, the percentage of neurons capable of generating action potentials decreased in the ALS lines. By seven weeks, many of those neurons fell silent, mustering no spike at all. This likely explains the conflict between the earlier papers, agreed experts who spoke with Alzforum. “[This paper] gives probably the best evidence to date for the idea of progression from hyperexcitability to reduced excitation,” said Brian Wainger of Massachusetts General Hospital in Charlestown, a member of the group that previously observed hyperexcitability.
It is hard to say how that weeks-long progression in the dish compares to the decades-long development of ALS in a person, said Robert Baloh of Cedars-Sinai Medical Center in Los Angeles, who saw excitability drop in ALS motor neurons. The events Devlin and others observed might all happen during the development of the human embryo, he speculated. Therefore, researchers cannot be sure how relevant these excitability changes are to human disease. That said, several studies in mouse models and people with ALS back up the idea that excitability goes up or down as the disease progresses (Bories et al., 2007; Kanai et al., 2006; see also Related Papers below).
What causes this up-and-down excitability? Miles was buoyed to see the same phenomenon in the TDP-43 and C9ORF72 lines. The two mutations could instigate a common pathway, perhaps via their effects on RNA processing, he speculated. Miles plans to explore in detail how excitability changes.
If hyperexcitability does turn out to be part of the ALS process, then Wainger already has a treatment in mind. In his previous work with the approved epilepsy drug Retigabine, a voltage-gated potassium channel agonist, he found that it blocks hyperexcitability and improves survival in ALS motor neurons. He is planning a Phase 2 trial to see if this medication affects motor neuron excitability in people with ALS.—Amber Dance
References
Series Citations
Paper Citations
- Sareen D, O'Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH. Targeting RNA Foci in iPSC-Derived Motor Neurons from ALS Patients with a C9ORF72 Repeat Expansion. Sci Transl Med. 2013 Oct 23;5(208):208ra149. PubMed.
- Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH Jr, Cudkowicz ME, Bean BP, Eggan K, Woolf CJ. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014 Apr 10;7(1):1-11. Epub 2014 Apr 3 PubMed.
- Bories C, Amendola J, Lamotte d'Incamps B, Durand J. Early electrophysiological abnormalities in lumbar motoneurons in a transgenic mouse model of amyotrophic lateral sclerosis. Eur J Neurosci. 2007 Jan;25(2):451-9. PubMed.
- Kanai K, Kuwabara S, Misawa S, Tamura N, Ogawara K, Nakata M, Sawai S, Hattori T, Bostock H. Altered axonal excitability properties in amyotrophic lateral sclerosis: impaired potassium channel function related to disease stage. Brain. 2006 Apr;129(Pt 4):953-62. Epub 2006 Feb 8 PubMed.
Further Reading
Papers
- Kuo JJ, Siddique T, Fu R, Heckman CJ. Increased persistent Na(+) current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol. 2005 Mar 15;563(Pt 3):843-54. PubMed.
- Zona C, Pieri M, Carunchio I. Voltage-dependent sodium channels in spinal cord motor neurons display rapid recovery from fast inactivation in a mouse model of amyotrophic lateral sclerosis. J Neurophysiol. 2006 Dec;96(6):3314-22. PubMed.
- Fuchs A, Kutterer S, Mühling T, Duda J, Schütz B, Liss B, Keller BU, Roeper J. Selective mitochondrial Ca2+ uptake deficit in disease endstage vulnerable motoneurons of the SOD1G93A mouse model of amyotrophic lateral sclerosis. J Physiol. 2013 Mar 18; PubMed.
- van Zundert B, Peuscher MH, Hynynen M, Chen A, Neve RL, Brown RH, Constantine-Paton M, Bellingham MC. Neonatal neuronal circuitry shows hyperexcitable disturbance in a mouse model of the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Neurosci. 2008 Oct 22;28(43):10864-74. PubMed.
- Quinlan KA, Schuster JE, Fu R, Siddique T, Heckman CJ. Altered postnatal maturation of electrical properties in spinal motoneurons in a mouse model of amyotrophic lateral sclerosis. J Physiol. 2011 May 1;589(Pt 9):2245-60. Epub 2011 Feb 28 PubMed.
- Vucic S, Nicholson GA, Kiernan MC. Cortical hyperexcitability may precede the onset of familial amyotrophic lateral sclerosis. Brain. 2008 Jun;131(Pt 6):1540-50. Epub 2008 May 9 PubMed.
News
- Blocking Glial Receptor Protects Motor Neurons in Culture and Mice
- Surprise Save: Excitability Protects Neurons from Lou Gehrig’s
- Endoplasmic Reticulum Protein Protects Motor Neurons from ALS
- Earliest ALS Defects Said to Start in Disparate Places
- Spinal Interneurons as Instigators of Excitotoxicity in ALS
Primary Papers
- Devlin AC, Burr K, Borooah S, Foster JD, Cleary EM, Geti I, Vallier L, Shaw CE, Chandran S, Miles GB. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat Commun. 2015 Jan 12;6:5999. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
I think that this is an exciting paper that reconciles the ALS motor neuron hyperexcitability/hypoexcitability dilemma. First, the current study reinforces that hyperexcitability is an intrinsic property of ALS motor neurons. More importantly, this initial hyperexcitability may have led to loss of normal neuronal function (loss of synaptic activity and action potential output), and suggests an exciting possibility: Would ALS motor neurons be protected if normal neuronal function was restored? Another recent study by Wainger et al. suggests that this might hold true (see Wainger et al., 2014). In vitro, the potassium channel activator Retigabine corrects the hyperexcitability phenotype of SOD1(A4V) motor neurons, and improves motor neuron survival.
More interestingly, the hyperexcitable property of motor neurons appears to be a convergent point for ALS, regardless of the disease-causing mutations. Although Devlin et al. speculate that aberrant RNA processing may contribute to motor neuron hyperexcitability, future work that elucidates the mechanistic links between the various ALS mutations and neuronal hyperexcitability would be a big step forward to help identify better drugs to slow down the progression of this motor neuron disease.
References:
Wainger BJ, Kiskinis E, Mellin C, Wiskow O, Han SS, Sandoe J, Perez NP, Williams LA, Lee S, Boulting G, Berry JD, Brown RH Jr, Cudkowicz ME, Bean BP, Eggan K, Woolf CJ. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014 Apr 10;7(1):1-11. Epub 2014 Apr 3 PubMed.
View all comments by Shi-Yan NgMake a Comment
To make a comment you must login or register.