Cholesterol Greases the Aβ Aggregation Machine
Quick Links
When left to their own devices, Aβ42 monomers tend to ignore each other, but add a little bit of the social lubricant into the mix, and the peptides hook up and form fibrils. So claims a May 7 paper in Nature Chemistry. Researchers led by Michele Vendruscolo at the University of Cambridge in England reported that exposure to vesicles embedded with cholesterol hastened the formation of Aβ42 fibrils in solution. Specifically, cholesterol accelerated primary nucleation—the oligomerization of Aβ42 monomers—as opposed to fueling the elongation of fibrils or the secondary seeding of new aggregates by the fibrils themselves.
- Cholesterol-laden vesicles accelerated Aβ42 aggregation in vitro.
- Fibrils formed in the presence of cholesterol were structurally similar to those formed without cholesterol.
- Cholesterol sped up nucleation of Aβ42 monomers into oligomers.
The researchers proposed that this catalysis could explain why Aβ42 aggregates in the brain, despite existing at very low concentrations there. The findings also offer mechanistic insight into known links between cholesterol and AD, and point to potential therapeutic strategies, Vendruscolo contended.
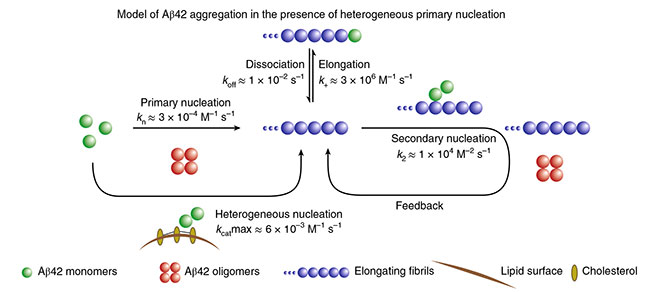
Cholesterol Catalysis. Aβ42 monomers (green) undergo primary nucleation into oligomers (red); this process accelerates in the presence of cholesterol-laden membranes (brown). After fibril elongation, secondary nucleation dominates. [Courtesy of Habchi et al., Nature Chemistry, 2018.]
Cholesterol is a crucial component of cellular membranes. It holds them together and facilitates interactions between membrane proteins. However, elevated plasma cholesterol stiffens arteries, causes cardiovascular disease, and associates with increased risk of AD. While myriad hypotheses exist to try to explain cholesterol’s influence on AD, the sterol’s association with Aβ has emerged as a prime candidate (Wood et al., 2014 ). Cholesterol has been spotted within Aβ plaques, and reportedly hastens APP processing by directly associating with APP C-terminal fragments in the membrane that harbor Aβ (Mori et al., 2001; Gellermann et al., 2005; Jun 2012 news). Researchers have reported that cholesterol facilitates Aβ oligomerization and the formation of amyloid pores in lipid bilayers, which damage cellular membranes (Di Scala et al., 2014; Ligouri et al., 2013).
In this study, co-first authors Johnny Habchi, Sean Chia, and Céline Galvagnion took a chemical kinetic approach to investigate cholesterol’s sway over Aβ aggregation. They started by mixing Aβ with membrane vesicles containing a mixture of phospholipids and cholesterol. Phosphatidylcholine is the most abundant phospholipid in neuronal membranes, so the researchers considered using it to form the basis of their vesicles. However, they found that the highly unsaturated nature of phosphatidylcholine’s hydrocarbon chains also influenced Aβ42 aggregation. To focus solely on the role of cholesterol in Aβ aggregation, they therefore used 1,2-dimyristoyl-sn-glycero-3-PC (DMPC), a form of phosphatidylcholine with more saturated chains. They then titrated varying levels of cholesterol into the DMPC vesicles, and added them to Aβ42 monomers.
Aggregation of Aβ42 accelerated as they increased cholesterol in the vesicles from 5 to 15 percent, but then the rate leveled off. The researchers attributed this threshold to cholesterol concentrating in clusters called lipid rafts. As additional cholesterol is added, it joins the rafts, without increasing the proportion of cholesterol throughout the rest of the lipid bilayer.
The researchers used circular dichroism to monitor the level of β-sheet structures in their vesicle/Aβ42 mixtures. In agreement with their fibrillization study, they found that β-sheets appeared more rapidly when Aβ42 was mixed with vesicles that contained 15 percent cholesterol than when it was mixed with vesicles that contained DMPC alone.
Did cholesterol affect the size or shape of Aβ fibrils that formed? To find out, the researchers used microscopic and structural techniques, including atomic force microscopy, cryo-electron microscopy, and Fourier-transform infrared spectroscopy. Overall, they found that Aβ42 fibrils in the test tube were similar in size and shape, regardless of whether they formed in the presence or absence of vesicles, or whether those vesicles contained 15 percent cholesterol or just DMPC.
How does cholesterol accelerate Aβ aggregation? Tracking changes in Thioflavin T fluorescence, the researchers found that cholesterol-laden membranes hastened the initiation of aggregation, but not the elongation of fibrils. This initiation step, dubbed primary nucleation, occurs when Aβ monomers form multimers. Through a series of kinetic modeling experiments, the researchers determined that cholesterol only enhances this step, and it does so by as much as 20-fold. Adding bexarotene, an inhibitor of primary nucleation, equally suppressed Aβ aggregation regardless of the presence of cholesterol. While previous studies have reported that fibrils can catalyze a far more efficient secondary nucleation process, cholesterol had no effect on this type of oligomerization (see image above).
Vendruscolo told Alzforum that cholesterol might make it possible for scarce Aβ42 peptides in the brain to condense and oligomerize. The kinetic experiments do not clarify how Aβ and cholesterol interact, but Vendruscolo hypothesized that the sterol might increase the local concentration of the peptide in the cell membrane enough to trigger aggregation.
Charles Sanders of Vanderbilt University in Nashville praised the thorough quantitative nature of the study. While it does not explain exactly how cholesterol affects Aβ aggregation, it does provide a solid kinetic understanding. “We know that something triggers the aggregation of Aβ peptides in the brain, and this paper characterizes one of the possible processes in great detail,” Sanders said. He previously reported that cholesterol interacts with the C99 fragment of APP, which is a cleavage product of BACE and a substrate for γ-secretase. The cholesterol bound to a site within the Aβ peptide sequence. It is possible that Aβ already has cholesterol bound to it when it is released, Sanders said. If so, that would provide a mechanistic link between his and Vendruscolo’s studies.
Tobias Hartmann of Saarland University in Homburg, Germany, also drew connections to previous studies. “Habchi et al. now show that cholesterol can drastically hasten Aβ42 aggregation. Not any aggregation, but quite specifically a primary heterologous Aβ42 nucleation process that leads to Aβ42 oligomers,” he wrote to Alzforum. “As elegant as is the experimental design and as welcome as is the clarity of the results, one needs to keep in mind that these are synthetic molecules in an artificial setting,” he cautioned. “Neither the membrane, nor the Aβ peptide composition and concentration match the incredibly more complex situation present in neurons or even more so that of a living brain.” Despite these caveats, Hartmann was fascinated by the implication that cholesterol-mediated aggregation of Aβ could play an early role in AD pathogenesis.
Vendruscolo acknowledged that compared with the highly complex and dynamic lipid milieu in the brain, the DMPC/cholesterol mixtures used in his study were highly simplistic. He also pointed out that molecules other than cholesterol also likely influence Aβ aggregation in the brain. Still, he said the study’s main finding is that under certain conditions, cholesterol accelerates Aβ aggregation. How often those conditions occur in a physiological setting, and whether they are influenced by poor cholesterol homeostasis, remains to be established, he said.
Sanders agreed, pointing out that there are myriad ways in which cholesterol might influence AD risk, including vascular disease, inflammation, and oxidative damage. Nonetheless, that some AD risk factors, such as ApoE, TREM2, and clusterin, affect cholesterol metabolism point to an important role for the sterol in AD. “The link between these genes and AD may indeed go through cholesterol,” Vendruscolo said.
Dieter Willbold of the Institute of Structural Biology and Biophysics in Juelich, Germany, also highlighted how the study connected different aspects of the disease process. “This technologically elegant study strengthens evidence for the role of Aβ aggregation in Alzheimer’s disease development by linking Aβ primary nucleation probability with cholesterol levels and with ApoE4 as a genetic risk factor.”—Jessica Shugart
References
News Citations
Paper Citations
- Wood WG, Li L, Müller WE, Eckert GP. Cholesterol as a causative factor in Alzheimer's disease: a debatable hypothesis. J Neurochem. 2014 May;129(4):559-72. Epub 2014 Jan 2 PubMed.
- Mori T, Paris D, Town T, Rojiani AM, Sparks DL, Delledonne A, Crawford F, Abdullah LI, Humphrey JA, Dickson DW, Mullan MJ. Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APP(SW) mice. J Neuropathol Exp Neurol. 2001 Aug;60(8):778-85. PubMed.
- Gellermann GP, Appel TR, Tannert A, Radestock A, Hortschansky P, Schroeckh V, Leisner C, Lütkepohl T, Shtrasburg S, Röcken C, Pras M, Linke RP, Diekmann S, Fändrich M. Raft lipids as common components of human extracellular amyloid fibrils. Proc Natl Acad Sci U S A. 2005 May 3;102(18):6297-302. PubMed.
- Di Scala C, Chahinian H, Yahi N, Garmy N, Fantini J. Interaction of Alzheimer's β-amyloid peptides with cholesterol: mechanistic insights into amyloid pore formation. Biochemistry. 2014 Jul 22;53(28):4489-502. Epub 2014 Jul 11 PubMed.
Further Reading
Papers
- Hellstrand E, Sparr E, Linse S. Retardation of Abeta fibril formation by phospholipid vesicles depends on membrane phase behavior. Biophys J. 2010 May 19;98(10):2206-14. PubMed.
- Popp J, Meichsner S, Kölsch H, Lewczuk P, Maier W, Kornhuber J, Jessen F, Lütjohann D. Cerebral and extracerebral cholesterol metabolism and CSF markers of Alzheimer's disease. Biochem Pharmacol. 2013 Jan 3; PubMed.
- Ghribi O, Larsen B, Schrag M, Herman MM. High cholesterol content in neurons increases BACE, beta-amyloid, and phosphorylated tau levels in rabbit hippocampus. Exp Neurol. 2006 Aug;200(2):460-7. PubMed.
- Di Scala C, Chahinian H, Yahi N, Garmy N, Fantini J. Interaction of Alzheimer's β-amyloid peptides with cholesterol: mechanistic insights into amyloid pore formation. Biochemistry. 2014 Jul 22;53(28):4489-502. Epub 2014 Jul 11 PubMed.
Primary Papers
- Habchi J, Chia S, Galvagnion C, Michaels TC, Bellaiche MM, Ruggeri FS, Sanguanini M, Idini I, Kumita JR, Sparr E, Linse S, Dobson CM, Knowles TP, Vendruscolo M. Cholesterol catalyses Aβ42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat Chem. 2018 Jun;10(6):673-683. Epub 2018 May 7 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of the Saarland
These are very helpful results to better understand how cholesterol may impact formation of toxic oligomers. We know about many links between cholesterol and AD, most of which support a risk-increasing role.
Cholesterol is extremely abundant in the human brain. So abundant that it dwarfs all other lipid species. Handling this vast amount of a single molecule brings its own problems. This only aggravates with the onset of AD neurodegeneration, when suddenly scores of membrane chunks, rich in cholesterol, become dispensable. Yet brain cholesterol catabolism has its own rules and strict limitations, which don’t apply elsewhere in the human body. What happens if the brain cholesterol level increases? One especially curious finding is that Aβ actually has a cholesterol binding motif, suggesting, among other implications, that Aβ fibrils should interact with cholesterol. Habchi et al. now show that cholesterol can drastically hasten Aβ42 aggregation. Not any aggregation, but quite specifically a primary heterologous Aβ42 nucleation process, that leads to Aβ42 oligomers. They didn’t observe any effect on secondary nucleation, which arguably produces the lion’s share of aggregated Aβ. Another impressive result is that the shape of the Aβ42 fibrils in AFM and EM images appears to very similar, irrespective of whether they were produced in presence or absence of cholesterol. This may indicate that the biological properties of cholesterol catalyzed Aβ42 oligomers may not be that different from the ones produced in absence of cholesterol. This is probably welcome news to anyone who studies Aβ42 toxicity.
As elegant as is the experimental design and as welcome as is the clarity of the results, one needs to keep in mind that these are synthetic molecules in an artificial setting. Neither the membrane, nor the Aβ peptide composition and concentration match the incredibly more complex situation present in neurons or even more so that of a living brain. Having said this, I can’t resist but be fascinated to see that Habchi’s research points towards cholesterol-mediated aggregation having an impact rather early in AD pathogenesis, which is where observational studies seem to typically put it.
St. Michael's Neurology and Pain Medicine
Cholesterol and amyloid accumulation. The known unknowns of a dangerous liaison.
These findings by Habchi et al. are most interesting. Although politics should be kept outside of the interpretation of scientific data, in 2002, Donald Rumsfeld, the then U.S. Secretary of State for Defense, said: “There are known knowns. There are things we know that we know. There are known unknowns. That is to say, there are things that we now know we don't know. But there are also unknown unknowns.” Paraphrasing David Logan (Logan, 2009), there are many known knowns in biology; however, the number of known unknowns is always far above and beyond the knowns in most fields. As such, there are a few known knowns about cholesterol and AD, but a lot more of known unknowns.
One known known is the observation that, in animals, a hypercholesterolemic diet leads to increased amyloid deposition (Refolo et al., 2000). However, such clear-cut observations in animals are not readily translated to the human brain, where one finds more complex interactions between cholesterol and amyloid (Pappolla et al., 2003).
It is also known that midlife hypercholesterolemia is a risk factor for AD, as demonstrated in several epidemiological studies (Pappolla et al., 2003; Kivipelto et al., 2006; Kivipelto and Solomon, 2006; Kivipelto et al., 2002; Notkola et al., 1998). However, the interpretation of the published data is marred by (pseudo)controversy, with some investigators claiming a negative association between cholesterol levels in serum and AD (Mielke et al., 2005; Reitz et al., 2008). This controversy is more apparent than real, since most studies showing a positive correlation between high serum cholesterol and AD have examined cholesterol levels at midlife (cohorts' ages ranged from 40 to 59 years) and then correlated these levels to later development of dementia. In contrast, most negative reports only included participants of advanced ages. Autopsy observations are in sync with the positive epidemiological studies, that is, they show that high levels of serum cholesterol correlate with presence of amyloid deposition in human brain only in the youngest subjects but not in the older subjects (Pappolla et al., 2003). Similar observations confirming this age-related dynamic were more recently reported in 2017, using amyloid PET imaging in human subjects by at least two different groups of investigators (Gottesman et al., 2017; Vemuri et al., 2017).
A known unknown, then, revolves around the age-related factor (or factors) that acts in concert with cholesterol to drive up Alzheimer’s risk. Important observations by Suzana Petanceska (Petanceska et al., 2003), some time ago, showed that as cholesterol levels increase, so does the level of ApoE expression in the brain. However, we do not know whether such a phenomenon could be age-related. Since ApoE is a carrier of both, cholesterol and Aβ, the observations by Habchi et al. create the perfect storm for Aβ aggregation.
While elevations of serum cholesterol during mid-life are known to elevate the risk for developing AD later in life, what triggers the full-blown condition and the onset of dementia, later in life, remains a known unknown, or perhaps even worse, an unknown unknown.
References:
Logan DC. Known knowns, known unknowns, unknown unknowns and the propagation of scientific enquiry. J Exp Bot. 2009;60(3):712-4. PubMed.
Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000 Aug;7(4):321-31. PubMed.
Pappolla MA, Bryant-Thomas TK, Herbert D, Pacheco J, Fabra Garcia M, Manjon M, Girones X, Henry TL, Matsubara E, Zambon D, Wolozin B, Sano M, Cruz-Sanchez FF, Thal LJ, Petanceska SS, Refolo LM. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology. 2003 Jul 22;61(2):199-205. PubMed.
Kivipelto M, Solomon A, Blennow K, Olsson AG, Winblad B. The new cholesterol controversy - a little bit of history repeating?. Acta Neurol Scand Suppl. 2006;185:1-2. PubMed.
Kivipelto M, Solomon A. Cholesterol as a risk factor for Alzheimer's disease - epidemiological evidence. Acta Neurol Scand Suppl. 2006;185:50-7. PubMed.
Kivipelto M, Helkala EL, Laakso MP, Hänninen T, Hallikainen M, Alhainen K, Iivonen S, Mannermaa A, Tuomilehto J, Nissinen A, Soininen H. Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med. 2002 Aug 6;137(3):149-55. PubMed.
Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P, Tuomilehto J, Nissinen A. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer's disease. Neuroepidemiology. 1998;17(1):14-20. PubMed.
Mielke MM, Zandi PP, Sjögren M, Gustafson D, Ostling S, Steen B, Skoog I. High total cholesterol levels in late life associated with a reduced risk of dementia. Neurology. 2005 Apr 20; PubMed.
Reitz C, Tang MX, Manly J, Schupf N, Mayeux R, Luchsinger JA. Plasma lipid levels in the elderly are not associated with the risk of mild cognitive impairment. Dement Geriatr Cogn Disord. 2008;25(3):232-7. PubMed.
Gottesman RF, Schneider AL, Zhou Y, Coresh J, Green E, Gupta N, Knopman DS, Mintz A, Rahmim A, Sharrett AR, Wagenknecht LE, Wong DF, Mosley TH. Association Between Midlife Vascular Risk Factors and Estimated Brain Amyloid Deposition. JAMA. 2017 Apr 11;317(14):1443-1450. PubMed.
Vemuri P, Knopman DS, Lesnick TG, Przybelski SA, Mielke MM, Graff-Radford J, Murray ME, Roberts RO, Vassilaki M, Lowe VJ, Machulda MM, Jones DT, Petersen RC, Jack CR Jr. Evaluation of Amyloid Protective Factors and Alzheimer Disease Neurodegeneration Protective Factors in Elderly Individuals. JAMA Neurol. 2017 Jun 1;74(6):718-726. PubMed.
Petanceska SS, DeRosa S, Sharma A, Diaz N, Duff K, Tint SG, Refolo LM, Pappolla M. Changes in apolipoprotein E expression in response to dietary and pharmacological modulation of cholesterol. J Mol Neurosci. 2003;20(3):395-406. PubMed.
Make a Comment
To make a comment you must login or register.