Can’t Degrade That Pesky Misfolded Protein? Push It Off the MAPS
Quick Links
When intracellular trash disposals overflow with misfolded proteins, cells may dump the excess onto their neighbors’ doorsteps. In the June 13 Nature Cell Biology, researchers introduced a secretion pathway that spews misfolded cytoplasmic proteins into the extracellular abyss by way of a cellular stress response. A chaperone called USP19 escorts the waste into a newly described export lane, particularly when the proteasome becomes overwhelmed. In a first hint toward relevance to neurodegeneration, the researchers, led by Yihong Ye of the National Institutes of Health in Bethesda, Maryland, reported that the chaperone-turned-bouncer dispatched α-synuclein in this manner. It is not known if neurons do this, too, yet the potential contribution of this secretion system to the spread of amyloidogenic proteins excited researchers.
“The findings add a new pathway to the list of potential therapeutic targets for neurodegenerative disease,” commented Lary Walker of Emory University in Atlanta, who was not involved in the work. “The study reaffirms that cells can respond to adversity in unexpected ways, and so it seems likely that more unconventional proteostatic pathways await discovery.” (See full comment below.)
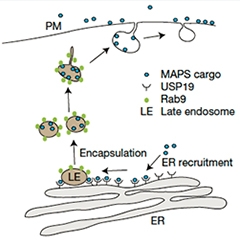
Toxic Dump?
In the MAPS pathway, USP19 recruits misfolded proteins to the ER membrane, where they are loaded into vesicles and shipped out to the cell surface. [Courtesy of Lee et al., Nature Cell Biology 2016.]
Cells expend a remarkable amount of energy disposing of misfolded proteins that threaten to gum up the works. The proteasome, which chomps up ubiquitinated proteins in the cytoplasm, is the best-known protein quality control system, while lysosomal degradation, autophagy, and chaperone-based unfolding and refolding also keep the cell tidy (see Wolff et al., 2014; Buchberger et al., 2010). Some misfolded proteins, including tau, reportedly leave the cell via tiny vesicular packages called exosomes; however, the researchers commented that export via exosomes tends to favor normal, folded proteins (see Saman et al., 2011; Lo Cicero et al., 2015).
Ye’s group was in the midst of investigating the role of USP19, the only deubiquitinating enzyme located in the ER membrane, in yet another protein quality-control pathway called ER-associated degradation (ERAD), when they stumbled across the new secretion system. In cells overexpressing the deubiquitinase, the researchers noticed an abundance of GFP, which was expressed as a cytosolic protein, floating in the cell medium. “This made me wonder how a cytosolic protein could be secreted,” Ye said.
First author Jin-Gu Lee and colleagues determined that HEK293T cells overexpressing USP19 secreted GFP in a manner dependent on USP19’s catalytic activity and localization to the ER membrane. GFP did not leave the cells because transfection made the cells leaky, as USP19 overexpression did not increase cellular permeability. Normal ER cargo such as clusterin, which get ferried through the ER to the Golgi and then the plasma membrane and contain signal sequences, were unaffected by USP19 overexpression, and blocking the normal ER secretion pathway with Brefeldin A did not affect USP19-mediated GFP secretion. These findings suggested to the researchers that the deubiquitinase was involved in an unconventional secretion pathway, which somehow targeted proteins in the cytoplasm rather than in the ER.
The researchers also noticed that the GFP secreted into the medium, which they detected using antibodies, had lost its fluorescence. Since fluorescence depends on the tight packaging of a fluorophore within the barrel-like structure of the protein, the researchers wondered if GFP had somehow lost its normal shape and that was why the cell dumped it. To investigate, they expressed a non-functional version of GFP (GFP1-10) that tends to misfold. The HEK cells secreted this wonky GFP even more than normal GFP, and this also depended upon USP19 catalytic activity.
What about proteins other than GFP? Out of a panel of highly expressed proteins, the researchers found that USP19 mediated the secretion of ubiquitin-like protein 4A (Ubl4A), but only when Ubl4A was overexpressed. Normally Ubl4A exists in a protein complex, so the researchers reasoned that the extra copies of lone Ubl4A were somehow targeted by the secretion pathway. At this point, they dubbed the new pathway “misfolding-associated protein secretion” (MAPS), owing to its preference for misfolded or unassembled proteins.
What about proteins implicated in neurodegenerative disease? Most of this paper—a massive 12-page study plus seven supplementary figures and videos—used the workhorse tools of GFP and HEK cells to nail down the basic biology of MAPS. In a first foray into neurodegeneration, the researchers found that α-synuclein, but not tau, was secreted in a USP19-dependent fashion. Several Parkinson’s disease- associated mutants of α-synuclein also hitched a ride out of the cell via MAPS. When the researchers knocked down USP19 using short interfering RNA, or wiped it out completely with CRISPR, they saw a dramatic drop in secretion of GFP1-10, Ubl4A, and α-synuclein from cells overexpressing these proteins.
To determine the relationship between MAPS and the proteasomal degradation pathway, the researchers treated cells with the proteasome inhibitor MG-132. They found a build-up of GFP1-10 and Ubl4A in the cell, suggesting that these proteins are normally degraded by the proteasome. In cells overexpressing USP19, proteasome inhibition diverted more GFP1-10 and Ubl4A into the MAPS pathway. Interestingly, when the researchers compared the ubiquitination state of these proteins inside and outside of the cell, they found that the secreted versions were lacking ubiquitin adornments, while those that remained in the cell were ubiquitinated. This suggested that USP19 removes ubiquitin as a way to target proteins into MAPS. Finally, the researchers found that the backup secretion system made cells hardier: A higher percentage of USP19-expressing cells survived transient exposure to the proteasome inhibitor than cells lacking USP19 expression. This indicated that the MAPS pathway provides critical relief during times when the proteasome cannot keep up with the cell’s demand for degradation.
How did USP19 mediate the secretion of MAPS substrates? Because USP19 is anchored to the cytosolic side of the ER membrane via a transmembrane domain at its C-terminus, the researchers investigated whether MAPS substrates also ended up there. They found that USP19 overexpression elevated the level of GFP1-10 in membrane fractions of cells, and confocal microscopy revealed comingling of USP19, GFP1-10, and the ER marker PDI.

All Aboard the MAPS Train. Misfolding-prone GFP1-10 (green) mingles with USP19 (red) at the ER membrane. The localization patterns of both proteins trace similar paths (bottom panels). [Courtesy of Lee et al., Nature Cell Biology 2016.]
After the ER, where to next? Live cell imaging revealed ER-associated vesicular bubbles periodically moving away from the ER domain. These so-called “MAPS vesicles” were mobile and expressed Rab9, much like late endosomes. Importantly, once released from the cell, the contents of these MAPS vesicles was free, rather than encapsulated in persistent vesicles, like exosomes, as the secreted GFP1-10 did not sediment via ultracentrifugation the way exosomes do.
Ye compared the MAPS model to a train. “Imagine the ER as the platform of a train station, whereas Rab9-positive late endosomes are cars on a train transporting passengers to the cell exterior,” he said. “USP19 is a chaperone that brings passengers to the platform.”
The idea that misfolded, toxic proteins spread between neurons in a prion-like fashion in neurodegenerative diseases has gathered steam, though it has also been called into question because mechanisms of transmission or toxicity still have to be proven (see Apr 2016 webinar).
“By identifying a previously unknown process by which cells secrete misfolded cytosolic proteins into the extracellular space, Lee and colleagues have taken a compelling step toward meeting this challenge,” Walker told Alzforum. Patrik Brundin of the Van Andel Research Institute in Grand Rapids, Michigan, agreed. “These are very exciting findings that potentially will impact research into novel therapeutic targets for Parkinson’s disease and related α-synucleinopathies,” he wrote. “While the authors have mostly focused their studies on non-neuronal cells, the findings are very important and provide insight into a key molecular mechanism of high relevance to Parkinson’s pathogenesis.” (See full comment below.)
Natura Myeku of Columbia University in New York voiced a concern shared by all commentators as well as the researchers themselves: “The study is to be complimented for its significance and quality results, but it remains to be seen if this mechanism is present in cultured primary neurons or in vivo, and under normal expression of USP19.” That α-synuclein, but not tau, is a MAPS substrate even though both proteins are implicated in proteopathic spread highlights the need to study this in the brain, Myeku added (see full comment below). Ye told Alzforum that he wants to collaborate with neuroscience labs to test the importance of MAPS in neurons and in neurodegenerative disease.
Marc Diamond of the University of Texas Southwestern Medical Center in Dallas, who studies tau propagation, commented that his lab may look into MAPS as a mechanism. About tau, he noted that Ye’s study did not investigate whether oligomers could be secreted via MAPS, noting that tau trimers, not misfolded monomers, are putative proteopathic seeds (see Apr 2015 news). Ye speculated that because MAPS substrates must somehow translocate from the cytoplasm into the ER, it seems unlikely that large aggregates could enter the secretion pathway.
How might MAPS secretion compare to protein release in exosomes? Last year, Tsuneya Ikezu’s group at Boston University reported that microglia transmitted tau between neurons via these tiny vesicular packages (see Oct 2015 news). Ikezu told Alzforum that while MAPS could be a bona fide pathway worthy of further investigation, the authors did not definitively rule out exosomal secretion of GFP1-10. While they saw no GFP1-10 in a would-be exosomal fraction, they had no positive control to prove that they could detect exosomes in the first place, Ikezu said. He added that the contribution of MAPS could vary between different cell types and between different isoforms of tau.
All researchers contacted by Alzforum agreed that replication and translation of the findings to neurons was a crucial next step. At the same time, they were enthusiastic about the potential importance of the pathway in neurodegeneration. Walker added that MAPS could potentially flag the earliest stages of neurodegeneration. “The transition from proteasomal degradation to MAPS might leave biochemical signatures that could serve as biomarkers during the long, clinically silent period that precedes obvious signs and symptoms of disease,” he said.—Jessica Shugart
References
Webinar Citations
News Citations
- Tau Triple Threat: Do Trimers Make Bad Seeds?
- Deadly Delivery: Microglia May Traffic Tau Via Exosomes
Paper Citations
- Wolff S, Weissman JS, Dillin A. Differential scales of protein quality control. Cell. 2014 Mar 27;157(1):52-64. PubMed.
- Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010 Oct 22;40(2):238-52. PubMed.
- Saman S, Kim W, Raya M, Visnick Y, Miro S, Jackson B, McKee AC, Alvarez VE, Lee NC, Hall GF. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid (CSF) in early Alzheimer's Disease. J Biol Chem. 2011 Nov 4; PubMed.
- Lo Cicero A, Stahl PD, Raposo G. Extracellular vesicles shuffling intercellular messages: for good or for bad. Curr Opin Cell Biol. 2015 Aug;35:69-77. Epub 2015 May 19 PubMed.
Further Reading
Papers
- Fontaine SN, Zheng D, Sabbagh JJ, Martin MD, Chaput D, Darling A, Trotter JH, Stothert AR, Nordhues BA, Lussier A, Baker J, Shelton L, Kahn M, Blair LJ, Stevens SM Jr, Dickey CA. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016 Jul 15;35(14):1537-49. Epub 2016 Jun 3 PubMed.
Primary Papers
- Lee JG, Takahama S, Zhang G, Tomarev SI, Ye Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat Cell Biol. 2016 Jun 13; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University Maastricht
Cellular homeostasis is essential for cells, in particular neurons, to assure proper network function.
In addition to well-defined cellular safeguards such as ERAD, the ubiquitin-proteasome system (UPS), and autophagy, now a new UPS pathway with USP-19 as a pivotal regulator has been added for short-term protection of cells.
This impressive paper presents a lot of evidence for MAPS excluding alternative explanations such as autophagosomes and exosomes. Indeed when the UPS stops delivering, the MAPS pathway, already at the level of the endoplasmic reticulum, is very useful.
Of course follow-up questions can be raised, such as what is the predictive value for the in-vivo situation? This point was raised by Schipanski et al., 2014, where a mouse model for a serpinopathy called FENIB, aka familial encephalopathy with inclusion bodies, was crossbred with a mouse model with a flawed UPS that is obtainable via Jackson labs, JAX 008833, line #3413.
In fact, this very paper by Lee et al. opens new avenues for research toward specific therapies for conformational diseases such as tauopathies and many polyglutamine diseases.
References:
Schipanski A, Oberhauser F, Neumann M, Lange S, Szalay B, Krasemann S, van Leeuwen FW, Galliciotti G, Glatzel M. Lectin OS-9 delivers mutant neuroserpin to endoplasmic reticulum associated degradation in familial encephalopathy with neuroserpin inclusion bodies. Neurobiol Aging. 2014 Oct;35(10):2394-403. Epub 2014 Apr 8 PubMed.
Emory University
Beginning in the 1980s, experiments have shown that prions injected into the eye translocate with high spatial and temporal specificity to downstream components of the visual system (Buyukmihci et al., 1983; Fraser, 1982; Kimberlin et al., 1986; Liberski et al., 2012; Liberski et al., 1990; Scott et al., 1992). The most parsimonious explanation for this systematic pattern of regional involvement is that the prions spread by axonal transport and cell-to-cell transfer. Recent studies have found that other neurodegeneration-associated misfolded proteins, including α-synuclein, tau, and Aβ, also appear to spread through the connectome. All evidence points to prion-like seeds as the pathogenic agents, but how they translocate has been uncertain.
In a recent review, Walsh and Selkoe challenged the research community to clarify the cellular mechanisms that underlie the spread of misfolded proteins within the brain (Walsh and Selkoe, 2016; Apr 2016 webinar). By identifying a previously unknown process by which cells secrete misfolded cytosolic proteins into the extracellular space, Lee and colleagues have taken a compelling step toward meeting this challenge.
Moreover, in pinpointing USP19 as a key chaperone for the secretion of certain misfolded proteins, the findings add a new pathway to the list of potential therapeutic targets for neurodegenerative disease. The study reaffirms that cells can respond to adversity in unexpected ways, and so it seems likely that more unconventional proteostatic pathways await discovery.
Some other issues are raised by the discovery of misfolding-associated protein secretion (MAPS). Misfolded proteins are favored by the unconventional secretion pathway when proteasomes become incapable of efficient degradation. Proteasomal function decreases with age (Saez and Vilchez, 2014), particularly in postmitotic cells (Keller et al., 2002); the predicted increase in MAPS could explain why advancing age is a risk factor for synucleinopathy.
It would be useful to know if secreted seeds normally are taken up and degraded by local glial cells, and, if so, whether this process declines with age or under conditions of cellular stress. From the standpoint of organismic survival, it seems counterproductive for cells to share proteopathic seeds with other cells. However, by temporarily lowering the local intracellular concentration of misfolded proteins, MAPS may at least delay cell death and brain dysfunction. The transition from proteasomal degradation to MAPS might leave biochemical signatures that could serve as biomarkers during the long, clinically silent period that precedes obvious signs and symptoms of disease.
References:
Buyukmihci N, Goehring-Harmon F, Marsh RF. Neural pathogenesis of experimental scrapie after intraocular inoculation of hamsters. Exp Neurol. 1983 Aug;81(2):396-406. PubMed.
Fraser H. Neuronal spread of scrapie agent and targeting of lesions within the retino-tectal pathway. Nature. 1982 Jan 14;295(5845):149-50. PubMed.
Kimberlin RH, Walker CA. Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J Gen Virol. 1986 Feb;67 ( Pt 2):255-63. PubMed.
Liberski PP, Hainfellner JA, Sikorska B, Budka H. Prion protein (PrP) deposits in the tectum of experimental Gerstmann-Sträussler-Scheinker disease following intraocular inoculation. Folia Neuropathol. 2012;50(1):85-8. PubMed.
Liberski PP, Yanagihara R, Gibbs CJ Jr, Gajdusek DC. Spread of Creutzfeldt-Jakob disease virus along visual pathways after intraocular inoculation. Arch Virol. 1990;111(1-2):141-7. PubMed.
Scott JR, Davies D, Fraser H. Scrapie in the central nervous system: neuroanatomical spread of infection and Sinc control of pathogenesis. J Gen Virol. 1992 Jul;73 ( Pt 7):1637-44. PubMed.
Walsh DM, Selkoe DJ. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci. 2016 Apr;17(4):251-60. PubMed.
Saez I, Vilchez D. The Mechanistic Links Between Proteasome Activity, Aging and Age-related Diseases. Curr Genomics. 2014 Feb;15(1):38-51. PubMed.
Keller JN, Gee J, Ding Q. The proteasome in brain aging. Ageing Res Rev. 2002 Apr;1(2):279-93. PubMed.
Associate Director of the Van Andel Research Institute, and Director of the Center for Neurodegenerative Science
The investigators have identified a novel pathway that is active during secretion of misfolded α-synuclein to the extracellular space. These are very exciting findings that potentially will impact research into novel therapeutic targets for Parkinson¹s disease and related α-synucleinopathies. The concept that α-synuclein can act in a prion-like fashion in Parkinson¹s disease has gained widespread acceptance, but it has remained controversial how, and when, misfolded α-synuclein is secreted from neurons. While the authors have mostly focused their studies on non-neuronal cells, the findings are very important and provide insight into a key molecular mechanism of high relevance to Parkinson's pathogenesis.
Columbia University
The study by Lee et al. has uncovered a new coping mechanism when cells are faced with impaired proteasome degradation and subsequent overload of aberrant proteins. This pathway is distinct from the exosome pathway, whereby misfolded proteins are selectively secreted through ER-associated late endosomes.
The study is to be complimented for its significance and quality results, but it remains to be seen if this mechanism is present in cultured primary neurons or in vivo, and under normal expression of USP19. In that regard, the authors provide physiological relevance by showing that overexpression of USP19 promotes α-synuclein but not tau secretion, although both proteins are known to have unfolding and cell-to-cell propagation properties. Further research in relevant transgenic mouse models can help elucidate the therapeutic implication of this pathway in inhibiting the spreading pathology in neurodegenerative diseases.
Make a Comment
To make a comment you must login or register.