Biomarkers of Errant Endosome Spotted in Cerebrospinal Fluid
Quick Links
Does jammed-up protein recycling in neurons cause proteins to spill into the cerebrospinal fluid in Alzheimer’s disease? As reported in the November 25 Science Translational Medicine, scientists led by Scott Small at Columbia University, New York, identified 26 proteins that were released into the CSF when VPS35, the main protein of the retromer complex, was knocked out in the forebrains of mice. Lo and behold, among them were fragments of tau and of two BACE1 substrates, APLP1 and CHL1. The same fragments are found in the CSF of people with mild cognitive impairment and AD. The data indirectly link CSF tau to BACE1 activity, which may hint at a new mechanism of AD pathology.
- In mice, broken retromer generates tau, BACE substrates in CSF.
- The same protein fragments float in CSF of people with MCI, AD.
- Fragments may be markers for endosomal dysfunction in AD.
“Together these findings suggest a relationship between pathological changes in neuronal endosomal trafficking and formation of tau species linked to Alzheimer's,” Amy Pooler, now at Sangamo Therapeutics, San Francisco, told Alzforum.
VPS35 is a core component of the retromer complex, a protein machine that helps endosomes recycle cell surface proteins. Retromer-induced traffic jams that cause protein pileups have been implicated in AD pathogenesis (Small et al., 2017; Jun 2012 news). In fact, VPS35 missense mutations have been found in people with AD, and VPS35 levels were lower in brain areas vulnerable to AD (Vardarajan et al., 2012; Rovelet-Lecrux, et al., 2015; Small et al., 2005). Small wondered if cells might release proteins into the interstitial fluid when the retromer goes awry, and if those proteins might end up in the CSF and be detectable there.
To find out, first author Sabrina Simoes and colleagues knocked out VPS35 in forebrain neurons of wild-type mice, confirming its depletion with immunofluorescence and western blotting. According to Small, the mice looked and behaved normally but had some cognitive deficits, which his group will detail in an upcoming paper.
The forebrain VPS35 knockouts had about 50 protein and protein fragment changes in their CSF compared to controls. Tsuneya Ikezu, Boston University, wanted to know if the detected changes were actually due to deleting VPS35 in the neurons. “I do not question the strong VPS35 knockdown in neurons, but there is a gap between the altered neurons and CSF,” he told Alzforum.
The researchers first focused on fragments from the BACE1 substrates APLP1 and CHL1. Their presence was not a surprise. While trapped in dysfunctional endosomes, BACE1 has more time to process its substrates, hence they build up. In fact, VPS35 deficiency has already been linked to increased BACE1 activity in mice and elevated APLP1 has been reported in the CSF of people with AD (Bhalla et al., 2012; Wang et al., 2012; Begcevic et al., 2018).
APLP1 and CHL1 did not seem randomly elevated; they seemed tied at the hip. In CSF from wild-type mice, fragment levels of both proteins tracked together—more of one also meant more of the other. The same was true in the forebrain VPS35 knockouts, but these mice had higher concentrations of both. This close relationship suggests that the two proteins are processed through the same pathway.
Intriguingly, the authors also spotted more tau in the CSF of the forebrain VPS35 knockouts. They found its mid-domain fragment, the most abundant fragment of tau in the CSF of people with AD (Barthélemy et al., 2016). Why would that be? BACE1 does not process tau.
Echoing this proposed link between tau and a functioning retromer, recent studies reported that removing VPS35 caused tau to build up, whereas overexpressing VPS35 mopped up extraneous tau (Carosi et al., 2020; Vagnozzi et al., 2019).
Could tau and BACE1 be related? To look for evidence of BACE activity in human CSF, Simoes and colleagues created a single-molecule array (SIMOA) assay to detect APLP1 and CHL1 fragments. The researchers scanned CSF samples from 40 cognitively normal people without amyloid plaques, 21 people with mild cognitive impairment (MCI), and 316 people with mild to moderate AD dementia. Again, APLP1 and CHL1 tracked together in all samples. AD and MCI CSF contained more of both proteins, potentially indicating a retromer-dependent endosomal blockage.
Alas, the relationships between tau and the two BACE1 substrates were more complicated. As expected, levels of all three tracked together in control CSF samples. However, successively higher levels of CSF tau in people with MCI, when plotted against their APLP1 and CHL1, formed a curve that rose, then dropped (see image below). Given that APLP1 sticks to amyloid plaques (Bayer et al., 1999), the researchers normalized the MCI samples based on each person’s Aβ42 level. Indeed, this revealed a linear relationship between tau and both BACE substrates. The researchers suspected that more tau meant a higher plaque load, which would trap more APLP1 and CHL1, essentially hiding them from the CSF assay.
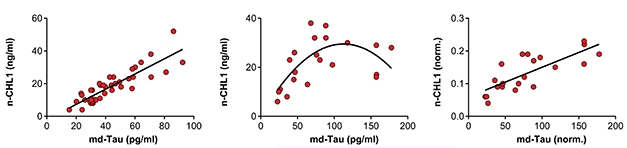
Plaques Mask Relationship. CSF tau and APLP1 were correlated in control CSF (left) but had a curved relationship in MCI samples (middle). After correcting for CSF Aβ42 levels, a linear relationship emerged (right). The same trends were true for tau and CHL1. [Courtesy of Simoes et al., Science Translational Medicine, 2020.]
The scientists then split the clinically diagnosed AD samples into those with and without amyloid plaques based on CSF Aβ42 levels. This revealed linear relationships between tau and APLP1 and between CHL1 in amyloid-positive CSF samples, but the correlations were tighter in amyloid-negative CSF samples.

Tight Trends. CSF APLP1, CHL1, and tau all correlated with one another in AD samples (top row). Samples with higher Aβ42 levels (red) had more of all three proteins than those without amyloid (blue, bottom rows). [Courtesy of Simoes et al., Science Translational Medicine, 2020.]
Henrik Zetterberg, University of Gothenburg, Sweden, found the correlation seen in AD CSF samples persuasive, including the differences in amyloid-positive and -negative samples. “This is exactly what you would expect to see: Aβ-positive individuals most likely have retromer dysfunction. This is how you could piece together the amyloid and retromer hypotheses,” he told Alzforum.
APLP1 and CHL1 correlated with phospho-tau217 (p-tau217), an early CSF biomarker specific to AD, albeit less tightly than they did with non-phosphorylated tau (Apr 2020 conference news; Barthélemy et al., 2020).
Taken together, endosomal trafficking—enabled by a well-oiled retromer—seems to be important to keep tau and BACE1 products out of the CSF. Retromer dysfunction in MCI and AD may be detectable by way of these proteins seeping into the CSF. This, then, connects CSF tau and BACE1 activity. Zetterberg said the work helps explain previous biomarker data showing that connection: “These data fit with what we have observed and not understood for many years.”
Thomas Karikari, also at UGothenburg, thinks the findings support the notion of tau changing early in AD pathology. “Specific p-tau markers arise almost concurrently with initial Aβ abnormalities, contrary to the commonly held theory that tau pathology begins several years after plaque pathology,” he told Alzforum (see full comment below).
Addressing more broadly how the retromer fits into the AD pathology continuum, Small proposes that dysfunctional endosome recycling lies upstream of all four AD pathologies: amyloid, tau, synaptic, and microglial. Therefore, he considers this, not amyloid accumulation, to be the event central in AD. “Amyloid pathology represents the disease's smoke, but not its fire,” he wrote together with Gregory Petsko in the same journal (Small and Petsko, 2020).
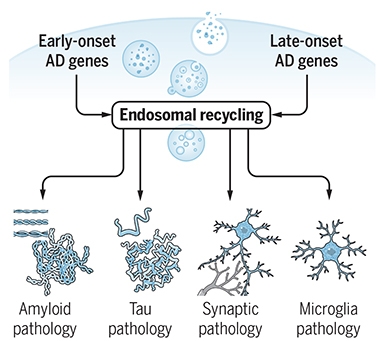
Central Problem? Mishaps with endosomal recycling contribute to all four Alzheimer’s pathologies, arguing for a key role in disease. [Courtesy of Scott and Petsko, Science Translational Medicine, 2020.]
Scott hopes the combination of elevated APLP1, CHL1, and tau will serve as biomarkers for endosomal dysfunction in AD. While thresholds for tau CSF levels are already established, Simoes is currently searching for a threshold to draw between normal and abnormal APLP1 and CHL1 levels.
Zetterberg suggested eventually using these biomarkers to evaluate drug candidates aimed at boosting retromer function. Like Ikezu, Pooler considers the new data promising, but both want to see studies in larger cohorts before these proteins can be called biomarkers.—Chelsea Weidman Burke
References
News Citations
- Coming Into Vogue? Retromer in APP Processing, AD Pathogenesis
- 217—The Best Phospho-Tau Marker for Alzheimer’s?
Paper Citations
- Small SA, Simoes-Spassov S, Mayeux R, Petsko GA. Endosomal Traffic Jams Represent a Pathogenic Hub and Therapeutic Target in Alzheimer's Disease. Trends Neurosci. 2017 Oct;40(10):592-602. PubMed.
- Vardarajan BN, Bruesegem SY, Harbour ME, St George-Hyslop P, Seaman MN, Farrer LA. Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol Aging. 2012 Sep;33(9):2231.e15-2231.e30. PubMed.
- Rovelet-Lecrux A, Charbonnier C, Wallon D, Nicolas G, Seaman MN, Pottier C, Breusegem SY, Mathur PP, Jenardhanan P, Le Guennec K, Mukadam AS, Quenez O, Coutant S, Rousseau S, Richard AC, Boland A, Deleuze JF, Frebourg T, Hannequin D, Campion D, CNR-MAJ collaborators. De novo deleterious genetic variations target a biological network centered on Aβ peptide in early-onset Alzheimer disease. Mol Psychiatry. 2015 Sep;20(9):1046-56. Epub 2015 Jul 21 PubMed.
- Small SA, Kent K, Pierce A, Leung C, Kang MS, Okada H, Honig L, Vonsattel JP, Kim TW. Model-guided microarray implicates the retromer complex in Alzheimer's disease. Ann Neurol. 2005 Dec;58(6):909-19. PubMed.
- Bhalla A, Vetanovetz CP, Morel E, Chamoun Z, Di Paolo G, Small SA. The location and trafficking routes of the neuronal retromer and its role in amyloid precursor protein transport. Neurobiol Dis. 2012 Jul;47(1):126-34. PubMed.
- Wang CL, Tang FL, Peng Y, Shen CY, Mei L, Xiong WC. VPS35 regulates developing mouse hippocampal neuronal morphogenesis by promoting retrograde trafficking of BACE1. Biol Open. 2012 Dec 15;1(12):1248-57. Epub 2012 Oct 11 PubMed.
- Begcevic I, Brinc D, Brown M, Martinez-Morillo E, Goldhardt O, Grimmer T, Magdolen V, Batruch I, Diamandis EP. Brain-related proteins as potential CSF biomarkers of Alzheimer's disease: A targeted mass spectrometry approach. J Proteomics. 2018 Jun 30;182:12-20. Epub 2018 Apr 22 PubMed.
- Barthélemy NR, Fenaille F, Hirtz C, Sergeant N, Schraen-Maschke S, Vialaret J, Buée L, Gabelle A, Junot C, Lehmann S, Becher F. Tau Protein Quantification in Human Cerebrospinal Fluid by Targeted Mass Spectrometry at High Sequence Coverage Provides Insights into Its Primary Structure Heterogeneity. J Proteome Res. 2016 Feb 5;15(2):667-76. Epub 2016 Jan 19 PubMed.
- Carosi JM, Hein LK, van den Hurk M, Adams R, Milky B, Singh S, Bardy C, Denton D, Kumar S, Sargeant TJ. Retromer regulates the lysosomal clearance of MAPT/tau. Autophagy. 2020 Sep 22;:1-21. PubMed.
- Vagnozzi AN, Li JG, Chiu J, Razmpour R, Warfield R, Ramirez SH, Praticò D. VPS35 regulates tau phosphorylation and neuropathology in tauopathy. Mol Psychiatry. 2019 Jul 9; PubMed. RETRACTED
- Bayer TA, Cappai R, Masters CL, Beyreuther K, Multhaup G. It all sticks together--the APP-related family of proteins and Alzheimer's disease. Mol Psychiatry. 1999 Nov;4(6):524-8. PubMed.
- Barthélemy NR, Bateman RJ, Hirtz C, Marin P, Becher F, Sato C, Gabelle A, Lehmann S. Cerebrospinal fluid phospho-tau T217 outperforms T181 as a biomarker for the differential diagnosis of Alzheimer's disease and PET amyloid-positive patient identification. Alzheimers Res Ther. 2020 Mar 17;12(1):26. PubMed.
- Small SA, Petsko GA. Endosomal recycling reconciles the Alzheimer's disease paradox. Sci Transl Med. 2020 Dec 2;12(572) PubMed.
Further Reading
Papers
- Zhang QY, Tan MS, Yu JT, Tan L. The Role of Retromer in Alzheimer's Disease. Mol Neurobiol. 2015 Jul 28; PubMed.
- Young JE, Fong LK, Frankowski H, Petsko GA, Small SA, Goldstein LS. Stabilizing the Retromer Complex in a Human Stem Cell Model of Alzheimer's Disease Reduces TAU Phosphorylation Independently of Amyloid Precursor Protein. Stem Cell Reports. 2018 Mar 13;10(3):1046-1058. Epub 2018 Mar 1 PubMed.
- Li JG, Chiu J, Praticò D. Full recovery of the Alzheimer's disease phenotype by gain of function of vacuolar protein sorting 35. Mol Psychiatry. 2019 Feb 7; PubMed. RETRACTED
Primary Papers
- Simoes S, Neufeld JL, Triana-Baltzer G, Moughadam S, Chen EI, Kothiya M, Qureshi YH, Patel V, Honig LS, Kolb H, Small SA. Tau and other proteins found in Alzheimer's disease spinal fluid are linked to retromer-mediated endosomal traffic in mice and humans. Sci Transl Med. 2020 Nov 25;12(571) PubMed.
- Small SA, Petsko GA. Endosomal recycling reconciles the Alzheimer's disease paradox. Sci Transl Med. 2020 Dec 2;12(572) PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Gothenburg
The authors report exciting new findings showing that two BACE1 substrates—the transmembrane proteins APLP1 and CHL1—are found, along with mid-domain tau, in the CSF of a mouse model of endosomal trafficking as well as in human AD versus control patient CSF.
The results build on previous findings on endosomal trafficking impairments in AD. To my mind, the most important contribution is the demonstration of associations between tau and BACE1 in endosomal trafficking, as well as the development and application of SIMOA assays to readily measure APLP1 and CHL1 levels in human and mouse CSF.
BACE1 acts on APP to initiate Aβ production and plaque formation, which, in turn, causes BACE1 accumulation in dystrophic neurites. Increased APLP1 and CHL1 could therefore be indicate plaque formation in AD. Elevation of total and phosphorylated tau in the same individuals, and their strong correlations with the BACE1 substrate levels, suggests that tau pathophysiological changes begin very early in the AD continuum, alongside Aβ abnormalities. This is in line with recent studies showing that, contrary to the commonly held theory that tau pathology begins several years after plaque pathology, specific p-tau variants begin to occur almost concurrently with initial Aβ abnormalities (Janelidze et al., 2020; Suárez-Calvet and Karikari et al., 2020; Karikari et al., 2020).
This close association between tau, Aβ, and BACE1 activity has been shown previously. Tau deletion in mouse models reduced plaque formation, plaque-associated BACE1 accumulation and APP accumulation around plaques (Peters et al., 2019). Pharmacological inhibition of BACE1 activity in APP transgenic mice reduced CSF tau and brain Aβ levels, suggesting that CSF tau can be used as an outcome measure for BACE1 inhibitor function (Schelle et al., 2016).
It will be interesting to compare the associations between different p-tau biomarkers (for example, p-tau181, p-tau217, p-tau231) with these BACE1-associated proteins in mouse models and humans.
References:
Janelidze S, Stomrud E, Smith R, Palmqvist S, Mattsson N, Airey DC, Proctor NK, Chai X, Shcherbinin S, Sims JR, Triana-Baltzer G, Theunis C, Slemmon R, Mercken M, Kolb H, Dage JL, Hansson O. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer's disease. Nat Commun. 2020 Apr 3;11(1):1683. PubMed.
Suárez-Calvet M, Karikari TK, Ashton NJ, Lantero Rodríguez J, Milà-Alomà M, Gispert JD, Salvadó G, Minguillon C, Fauria K, Shekari M, Grau-Rivera O, Arenaza-Urquijo EM, Sala-Vila A, Sánchez-Benavides G, González-de-Echávarri JM, Kollmorgen G, Stoops E, Vanmechelen E, Zetterberg H, Blennow K, Molinuevo JL, ALFA Study. Novel tau biomarkers phosphorylated at T181, T217 or T231 rise in the initial stages of the preclinical Alzheimer's continuum when only subtle changes in Aβ pathology are detected. EMBO Mol Med. 2020 Dec 7;12(12):e12921. Epub 2020 Nov 10 PubMed.
Karikari TK, Emeršič A, Vrillon A, Lantero-Rodriguez J, Ashton NJ, Kramberger MG, Dumurgier J, Hourregue C, Čučnik S, Brinkmalm G, Rot U, Zetterberg H, Paquet C, Blennow K. Head-to-head comparison of clinical performance of CSF phospho-tau T181 and T217 biomarkers for Alzheimer's disease diagnosis. Alzheimers Dement. 2021 May;17(5):755-767. Epub 2020 Nov 30 PubMed. Correction.
Peters F, Salihoglu H, Pratsch K, Herzog E, Pigoni M, Sgobio C, Lichtenthaler SF, Neumann U, Herms J. Tau deletion reduces plaque-associated BACE1 accumulation and decelerates plaque formation in a mouse model of Alzheimer's disease. EMBO J. 2019 Dec 2;38(23):e102345. Epub 2019 Nov 7 PubMed.
Schelle J, Häsler LM, Göpfert JC, Joos TO, Vanderstichele H, Stoops E, Mandelkow EM, Neumann U, Shimshek DR, Staufenbiel M, Jucker M, Kaeser SA. Prevention of tau increase in cerebrospinal fluid of APP transgenic mice suggests downstream effect of BACE1 inhibition. Alzheimers Dement. 2016 Oct 14; PubMed.
Make a Comment
To make a comment you must login or register.