APP Secretases Play Tug of War with Growth Cones: Could Inhibitors Dictate the Winner?
Quick Links
Like factory workers in a processing line, β- and γ-secretase make successive snips in amyloid precursor protein (APP), releasing Aβ peptides. Now, a study reports that this enzymatic duo also coordinates axon growth—a key factor in neural development and synaptic plasticity. As described in the August 20 Cell Reports, β-secretase (BACE1) activates a cell adhesion molecule called close homolog of L1 (CHL1), which then triggers growth cones on the tips of axons to collapse. These protrusions influence the direction of axonal growth. γ-Secretase then clips this activated form of CHL1, allowing the protrusions to grow again. These newfound roles for β- and γ-secretase raise concerns about off-target effects of BACE or γ-secretase inhibitors. However, the researchers, led by Bart De Strooper at KU Leuven in Belgium, reported that higher levels of BACE1 inhibitors than Aβ production are needed to affect growth cone dynamics.
“This is an excellent paper, which finally provides a molecular function and a mechanism for the cleavage of CHL1 by BACE1,” commented Stefan Lichtenthaler of the German Center for Neurodegenerative Diseases in Munich. Lichtenthaler and De Strooper independently identified CHL1 as a BACE1 substrate (see Kuhn et al., 2012; Zhou et al., 2012).
As co-conspirators in the production of Aβ, BACE1 and γ-secretase are among the prime targets for drugs to treat Alzheimer’s disease. In addition to chopping up APP, the enzymes have other substrates with potentially important physiological functions that could be compromised by inhibitors. A clinical trial that tested the γ-secretase inhibitor semagacestat ended prematurely due to side effects and lack of efficacy (see Feb 2015 news), however, BACE1 inhibitors are currently being tested in several clinical trials (see Apr 2015 conference news and Ghosh and Osswald, 2014). Dozens of BACE1 substrates have been identified, but evidence for the physiological relevance of each enzyme/substrate pair is lacking (see Jun 2012 news).

Repulsion Relief.
Axonal growth cones retract away from cells pumping out Sema3A (top). This repulsion is blocked in thalamic neurons lacking BACE1 or CHL1, or treated with a BACE1 inhibitor. [Courtesy of Barão et al., Nature 2015.]
De Strooper and colleagues had reason to believe that BACE cleavage of CHL1 has physiological importance. Animals lacking CHL1 share similar characteristics with BACE1 knockouts. Axons in the adult hippocampus and olfactory bulb are misguided in both, and the animals behaved abnormally in tests of anxiety and hyperactivity.
CHL1 is a transmembrane subunit of the semaphorin 3A (Sema3A) receptor. Sema3A, a secreted protein that repels probing axons, triggers the collapse of growth cones. Previous studies have reported that Sema3A hooks up with another subunit of the receptor, neuropilin 1 (NRP1), to recruit CHL1, but researchers did not understand how this event leads to subsequent retreat of axons (see Wright et al., 2007).
For the current study, first author Soraia Barão and colleagues teased apart the relationship between BACE1 and CHL1. The researchers knew Sema3A directs axonal pathfinding across thalamocortical circuits during development. They therefore measured growth cone collapse on cultured thalamic explants. As expected, growth cones on these thalamic axons retreated from a cellular source of Sema3A. Axons from either CHL1 or BACE1 knockout mice were unaffected by the semaphorin. When the researchers treated thalamic neurons with a stabilized form of Sema3A they observed axon-wide growth cone collapse. This did not occur in the presence of BACE1 inhibitors. These results indicated that BACE1, via CHL1, mediates the action of Sema3A on growth cones.
How did BACE1 mediate the collapse? The researchers found that Sema3A increased proteolytic shedding of CHL1 from the cell surface. Furthermore, when the researchers abolished the BACE1 cleavage site in CHL1, it was unable to restore growth cone defects when expressed in CHL1 knockouts.
Similarly to APP processing, BACE1 cleavage of CHL1 produces an N terminal fragment (NTFβ) that is shed from the membrane, and a C-terminal fragment (CTFβ) that stays put. Interestingly, expression of CTFβ, but not NTFβ, rescued the collapse defect in CHL1 knockouts, indicating that this cleavage fragment mediated the cone retreat. To find out how, the researchers mutated key regions in CHL1 that reside within the CTFβ fragment. They found that the ezrin, radixin, and moesin (ERM) domain was necessary for collapse. This domain binds ERM proteins, which facilitate actin depolymerization and growth cone retraction.
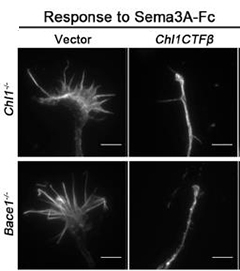
Fragment to the Rescue.
Growth cones in neurons lacking CHL1 or BACE1 hold their ground in response to Sema3A (left panel). The C-terminal, but not N-terminal, fragment of CHL1 rescues this defect (right panel). [Courtesy of Barão et al., Cell Reports, 2015.]
Lichtenthaler was surprised that the C-terminal fragment, rather than the shed ectodomain, was the pivotal mediator of growth cone collapse. Barão hypothesized that removal of the NTFβ fragment could allow ERM proteins to access CTFβ, but the idea has yet to be tested.
The data so far indicate that in response to Sema3A, BACE1 cleaves CHL1, which, through the ERM domain in its CTFβ fragment, leads to the collapse of growth cones. What restores the growth cone? That’s where γ-secretase comes in. Treatment of primary mixed brain neuronal cultures with γ-secretase inhibitors led to a build up of CHL1’s CTFβ fragment, indicating that γ-secretase normally chops it up. Blocking γ-secretase strongly enhanced growth cone collapse. Using knockout mice, the researchers determined that the B, but not the A, isoform of APH1, one of the γ-secretase subunits, was required for the recovery of cones after they collapse.
Live cell imaging experiments confirmed the role of γ-secretase. In neurons treated with a γ-secretase inhibitor, growth cone recovery failed to take place after washing out Sema3A. Instead, axons continued to retract their protrusions.
The researchers proposed a mechanism whereby engagement of CHL1 by Sema3A triggers the receptor’s internalization into endosomes, where BACE1 resides. BACE1 then cleaves CHL1 revealing the CTFβ fragment, which in turn triggers growth cone collapse by an as-yet-unknown mechanism. The collapse stops once γ-secretase cleaves CHL1 CTFβ—either within the endosomes or after recycling to the cell surface. This cascade mirrors the dynamism of axonal guidance, Barão told Alzforum, in which growth and retraction are simultaneous processes that respond constantly to external cues. “Growth cone collapse is a very transient event, because the axon has to collapse in a certain area while continuing to progress in the direction of the target,” she said.
Would BACE1 inhibitors, which are being tested in AD patients in clinical trials, interfere with growth cone dynamics in adults? To chip away at this question, the researchers measured the dose response for both growth cone collapse and production of Aβ. They found that while only 3 nM of the BACE1 inhibitor CIV was needed to cut Aβ40 production almost in half, 100-fold more was required to prevent Sema3A-induced growth cone collapse. This suggested that low doses of BACE1 inhibitors could reduce amyloid production without harming growth cone dynamics.
Commentators agreed that these results suggested an appropriate dose window could exist for treatment, although that idea remains to be tested. An equally pressing issue Barão and others raised was whether growth cone collapse plays an important role in the adult brain. Growth cones are essential during development, but their role after neural circuits have gelled is less clear. Barão said that the pathway could be important for maintaining circuitry and preventing axons from straying into inappropriate territory. In the hippocampus and olfactory bulb, where newborn neurons emerge throughout adulthood, axonal guidance pathways could remain crucial in assimilating these newbies, she added.
“The implications of these findings are that accurate dosing and careful monitoring of BACE1-dependent cleavage products in the CSF, and of olfactory and memory function, would be important in the analysis of clinical trials of BACE inhibitors,” commented Mark Albers of Massachusetts General Hospital in Charlestown. “Of course, the latter will be challenging since both olfaction and memory are compromised in untreated Alzheimer’s disease.”
Riqiang Yan of the Cleveland Clinic in Ohio was skeptical about whether this pathway was important beyond development. “In my opinion, this is another elegant demonstration of the important role of BACE1 in the control of neural development,” he wrote. “BACE1 inhibition is unlikely to cause severe side effects in elderly people due to the disruption of this particular pathway.”—Jessica Shugart
References
Therapeutics Citations
News Citations
- Semagacestat Failure Analysis: Should γ-Secretase Remain a Target?
- At AD/PD Meeting, New BACE Inhibitor Struts Its Stuff
- BACE Secrets: Newly Identified Substrates May Regulate Plasticity
Paper Citations
- Kuhn PH, Koroniak K, Hogl S, Colombo A, Zeitschel U, Willem M, Volbracht C, Schepers U, Imhof A, Hoffmeister A, Haass C, Rossner S, Bräse S, Lichtenthaler SF. Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 2012 Jul 18;31(14):3157-68. PubMed.
- Zhou L, Barão S, Laga M, Bockstael K, Borgers M, Gijsen H, Annaert W, Moechars D, Mercken M, Gevaert K, Gevaer K, De Strooper B. The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J Biol Chem. 2012 Jul 27;287(31):25927-40. PubMed.
- Ghosh AK, Osswald HL. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer's disease. Chem Soc Rev. 2014 Sep 8;43(19):6765-813. PubMed.
- Wright AG, Demyanenko GP, Powell A, Schachner M, Enriquez-Barreto L, Tran TS, Polleux F, Maness PF. Close homolog of L1 and neuropilin 1 mediate guidance of thalamocortical axons at the ventral telencephalon. J Neurosci. 2007 Dec 12;27(50):13667-79. PubMed.
Further Reading
Papers
- Vassar R, Kuhn PH, Haass C, Kennedy ME, Rajendran L, Wong PC, Lichtenthaler SF. Function, therapeutic potential and cell biology of BACE proteases: current status and future prospects. J Neurochem. 2014 Jul;130(1):4-28. Epub 2014 Apr 19 PubMed.
Primary Papers
- Barão S, Gärtner A, Leyva-Díaz E, Demyanenko G, Munck S, Vanhoutvin T, Zhou L, Schachner M, López-Bendito G, Maness PF, De Strooper B. Antagonistic Effects of BACE1 and APH1B-γ-Secretase Control Axonal Guidance by Regulating Growth Cone Collapse. Cell Rep. 2015 Sep 1;12(9):1367-76. Epub 2015 Aug 20 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.