Aggregates of TDP-43 in Muscle—Potential ALS Marker?
Quick Links
Could TDP-43 within the nerve bundles running in a person's muscles identify people who are developing amyotrophic lateral sclerosis (ALS)? Researchers led by Takashi Kurashige, Hiroshima University, Japan, think so. In the May 23 JAMA Neurology, they reported finding inclusions of the RNA-binding protein in muscle samples taken postmortem from people who had had sporadic ALS. Importantly, they also found TDP-43 aggregates in muscle biopsies from 33 of 36 people who had motor neuron symptoms and were later diagnosed with ALS. The findings suggest that for ALS, aggregates of TDP-43 in muscle tissue might be an early marker, something that has eluded scientists thus far.
- TDP-43 speckles axons within postmortem ALS muscle tissue.
- Muscle biopsies positive for TDP-43 predicted ALS.
- All TDP-43 positive people had motor symptoms per new diagnostic criteria.
Clinicians diagnose ALS based on deteriorating motor function, such as weakness in the arms or legs or difficulty walking. Motor neurons in the brain and spinal cord of people with ALS contain cytoplasmic inclusions of TDP-43, but currently, these can only be detected after a person has died (Oct 2006 news). Recently, other researchers had looked for TDP-43 in muscle biopsies from three cases, spotting inclusions within intramuscular nerve bundles (Altman et al., 2021).
To see if this held true in a larger sample, Kurashige, who is also first author on the paper, analyzed bicep, quadricep, diaphragm, or tongue muscles postmortem from 10 sporadic ALS cases and 12 age- and sex-matched controls. Within the tissue, he saw intramuscular nerves that had atrophied and puncta of phosphorylated TDP-43 in nerve axons (see image below). Neither occurred in the control tissue. Hyperphosphorylated TDP-43 forms aggregates in ALS and other neurodegenerative diseases. The findings suggest that TDP-43 inclusions within muscle tissue could become a marker for late-stage ALS.
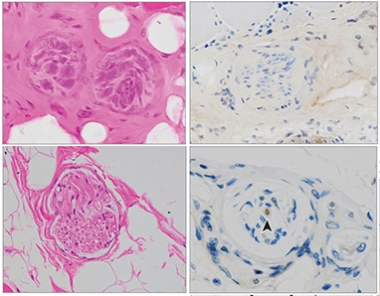
Shriveled, Speckled. Compared to muscle tissue taken from a healthy person (top), intramuscular nerve bundles from a person with ALS (bottom) appeared shrunken (left) and contained TDP-43 inclusions (arrowhead right). [Courtesy of Kurashige et al., JAMA Neurology, 2022.]
What about early stage disease? Kurashige analyzed muscle biopsies taken from adults who were experiencing muscle weakness. This minor surgical procedure involves slicing the skin to remove a small chunk of muscle, often from the thigh, bicep, or shoulder. Kurashige selected people whose biopsies had indicated no clear diagnosis of a neuromuscular disease, who had no family history of ALS or other neuromuscular disease, and who carried no known ALS mutations. Two-thirds of the participants were men, and their average age was 62.
Of the available biopsies from 114 participants, 71 had captured intramuscular nerve bundles. Of these samples, 33 contained TDP-43 inclusions in nerve axons. All 33 volunteers were subsequently diagnosed with ALS an average of five months after biopsy. Of the 38 participants whose biopsies contained nerve bundles but no TDP-43 inclusions, none developed ALS; rather, all were later diagnosed with a different neurodegenerative disease, such as spinal muscular atrophy. Three people whose biopsies had not captured nerve bundles also went on to develop ALS.
Of the 36 people who developed ALS, only nine were suspected of having it based on the commonly used Awaji criteria. This newer diagnostic incorporates electrophysiological data into the gold standard, the revised El Escorial criteria, which was based on a neurological exam (Jun 2010 news). Research suggests that the Awaji criteria are a bit more sensitive (Geevasinga et al., 2016). Still, both criteria are prone to error because clinicians must interpret clinical symptoms or electrophysiological signs of muscle atrophy as being definite, probable, or possible indicators of ALS, though all three mean the person has the disease (Braun et al., 2020; Johnsen et al., 2019).
To better identify ALS at the time of biopsy, Kurashige assessed clinical and electrophysiological data using the new “Gold Coast” criteria. These came about from a 2019 meeting in Australia, organized by the International Federation of Clinical Neurophysiology, the World Federation of Neurology, the ALS Association, and the MND Association. Clinicians simplified the process by diagnosing patients as having ALS, or not, based on straightforward clinical signs and/or electrophysiological data (Shefner et al., 2020). Once other motor diseases have been excluded, these new criteria diagnose ALS by the presence of progressive upper and lower motor dysfunction or only lower motor dysfunction in two or more muscle groups. Evidence suggests Gold Coast criteria are more sensitive than Awaji and pick up motor symptoms earlier in the disease process (Hannaford et al., 2021; Vucic et al., 2021; Shen et al., 2021).
In the Japanese study, 33 of the 36 people who would go on to have ALS fulfilled the Gold Coast criteria at the point of biopsy: 27 had upper and lower motor symptoms; six had only the latter in two or more regions. Interestingly, three people who fell below the Gold Coast diagnostic threshold because they had lower motor symptoms in only one region, had tested positive for intramuscular TDP-43 deposits on biopsy. All told, the authors think that pairing Gold Coast diagnostic criteria with biopsy tests for intramuscular TDP-43 inclusions could improve the diagnosis of ALS in its early stages.—Chelsea Weidman Burke
References
News Citations
- New Ubiquitinated Inclusion Body Protein Identified
- ALS: Out with El Escorial, In With Awaji-shima Trial Criteria
Paper Citations
- Altman T, Ionescu A, Ibraheem A, Priesmann D, Gradus-Pery T, Farberov L, Alexandra G, Shelestovich N, Dafinca R, Shomron N, Rage F, Talbot K, Ward ME, Dori A, Krüger M, Perlson E. Axonal TDP-43 condensates drive neuromuscular junction disruption through inhibition of local synthesis of nuclear encoded mitochondrial proteins. Nat Commun. 2021 Nov 25;12(1):6914. PubMed.
- Geevasinga N, Loy CT, Menon P, de Carvalho M, Swash M, Schrooten M, Van Damme P, Gawel M, Sonoo M, Higashihara M, Noto Y, Kuwabara S, Kiernan MC, Macaskill P, Vucic S. Awaji criteria improves the diagnostic sensitivity in amyotrophic lateral sclerosis: A systematic review using individual patient data. Clin Neurophysiol. 2016 Jul;127(7):2684-91. Epub 2016 Apr 16 PubMed.
- Braun N, Macklin EA, Sinani E, Sherman A, Weber M, Pooled Resource Open-Access ALS Clinical Trials Consortium. The revised El Escorial criteria "clinically probable laboratory supported ALS"-once a promising now a superfluous category?. Amyotroph Lateral Scler Frontotemporal Degener. 2020 Feb;21(1-2):24-28. Epub 2019 Sep 27 PubMed.
- Johnsen B, Pugdahl K, Fuglsang-Frederiksen A, Kollewe K, Paracka L, Dengler R, Camdessanché JP, Nix W, Liguori R, Schofield I, Maderna L, Czell D, Neuwirth C, Weber M, Drory VE, Abraham A, Swash M, de Carvalho M. Diagnostic criteria for amyotrophic lateral sclerosis: A multicentre study of inter-rater variation and sensitivity. Clin Neurophysiol. 2019 Feb;130(2):307-314. Epub 2018 Dec 9 PubMed.
- Shefner JM, Al-Chalabi A, Baker MR, Cui LY, de Carvalho M, Eisen A, Grosskreutz J, Hardiman O, Henderson R, Matamala JM, Mitsumoto H, Paulus W, Simon N, Swash M, Talbot K, Turner MR, Ugawa Y, van den Berg LH, Verdugo R, Vucic S, Kaji R, Burke D, Kiernan MC. A proposal for new diagnostic criteria for ALS. Clin Neurophysiol. 2020 Aug;131(8):1975-1978. Epub 2020 Apr 19 PubMed.
- Hannaford A, Pavey N, van den Bos M, Geevasinga N, Menon P, Shefner JM, Kiernan MC, Vucic S. Diagnostic Utility of Gold Coast Criteria in Amyotrophic Lateral Sclerosis. Ann Neurol. 2021 May;89(5):979-986. Epub 2021 Feb 24 PubMed.
- Vucic S, Ferguson TA, Cummings C, Hotchkin MT, Genge A, Glanzman R, Roet KC, Cudkowicz M, Kiernan MC. Gold Coast diagnostic criteria: Implications for ALS diagnosis and clinical trial enrollment. Muscle Nerve. 2021 Nov;64(5):532-537. Epub 2021 Aug 24 PubMed.
- Shen D, Yang X, Wang Y, He D, Sun X, Cai Z, Li J, Liu M, Cui L. The Gold Coast criteria increases the diagnostic sensitivity for amyotrophic lateral sclerosis in a Chinese population. Transl Neurodegener. 2021 Aug 9;10(1):28. PubMed.
Further Reading
Primary Papers
- Kurashige T, Morino H, Murao T, Izumi Y, Sugiura T, Kuraoka K, Kawakami H, Torii T, Maruyama H. TDP-43 Accumulation Within Intramuscular Nerve Bundles of Patients With Amyotrophic Lateral Sclerosis. JAMA Neurol. 2022 Jul 1;79(7):693-701. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Barrow Neurological Institute
In this recent paper, evaluation of intramuscular nerve bundles emerges as frequent site of TDP43 pathology. A postmortem case control study showed TDP43 accumulations in every specimen from ALS patients, and in no control cases. These accumulations appear more frequently in nerve bundles than in the CNS; whereas over 90 percent of motor neurons do not have accumulations in brain and spinal cord, about 50 percent of nerve muscle specimens show this pathology. The selectivity for ALS was confirmed in a retrospective analysis of open muscle biopsies.
Based on this study, muscle biopsy may again emerge as important diagnostic tool for patients in whom the diagnosis of ALS is viable but not established. These results also emphasize the potential importance of the nerve terminal and neuromuscular junction as targets for therapy.
Houston Methodist
Kurashige et al. found pTDP-43 in intramuscular nerve bundles in 10 of 10 autopsy-confirmed ALS patients, and none in 12 control patients without ALS. In a cohort study, 33 of 33 patients subsequently diagnosed with ALS had pTDP-43 in intramuscular nerve bundles, while zero of 38 individuals with pTDP-43–negative nerve bundles were subsequently diagnosed with ALS. On this basis, the authors conclude that pTDP-43-positive nerve bundles are specific for ALS and might be considered as a “novel diagnostic biomarker for ALS."
However, a paper published in Brain earlier this year (Riva et al., 2022) could not document such specificity, and would suggest that pTDP-43 positive intramuscular nerve biopsies would not be a reliable diagnostic biomarker for ALS. The data from Riva et al. was obtained from diagnostic motor nerve biopsies in 102 lower motor neuron syndrome patients. Axonal accumulation of pTDP43 was found in 98.2 percent of ALS cases, but was also found in 30.4 percent of non-ALS cases. Thus accumulation of TDP-43 is not specific for ALS and therefore would not be a meaningful diagnostic biomarker.
A possible explanation of the difference in the two studies may be that Kurashige was examining “intramuscular nerve bundle” specimens while Riva was examining “diagnostic motor nerve biopsies.” However, a lack of specificity is a more likely finding given the fact that pTDP-43 is not specific for ALS and has been reported in a wide range of tissues and conditions. Furthermore, biopsies of muscle nerves or intramuscular nerves are sufficiently invasive that they are unlikely to become readily accepted diagnostic procedures.
Nevertheless, both papers provide valuable documentation that TDP-43 is present long before a diagnosis of ALS can be made; the presence of pTDP-43 axonal aggregates may well be an early event in the pathogenesis of ALS, occurring well before axonal degeneration.
References:
Riva N, Gentile F, Cerri F, Gallia F, Podini P, Dina G, Falzone YM, Fazio R, Lunetta C, Calvo A, Logroscino G, Lauria G, Corbo M, Iannaccone S, Chiò A, Lazzerini A, Nobile-Orazio E, Filippi M, Quattrini A. Phosphorylated TDP-43 aggregates in peripheral motor nerves of patients with amyotrophic lateral sclerosis. Brain. 2022 Mar 29;145(1):276-284. PubMed.
Make a Comment
To make a comment you must login or register.