Not Just Blood Pressure—Dietary Salt Linked to Tau Phosphorylation
Quick Links
Too much salty food wreaks havoc on the cardiovascular system, raising blood pressure, damaging small blood vessels, and limiting perfusion into the brain. But is this why salt increases the chances of cognitive impairment? Not so fast. At this year’s Society for Neuroscience meeting, held October 19–23 in Chicago, Giuseppe Faraco from Costantino Iadecola’s lab at Feil Family Brain and Mind Research Institute of Weill Cornell Medicine, New York, reported that learning and memory deficits in mice chowing on a high-salt diet correlated with phosphorylation of tau, not with damage to the brain’s blood vessels. The study, published October 23 in Nature, links reduced nitric oxide in blood vessel walls to activation of kinases that modify tau. The findings present a new twist in the well-known link between cardiovascular disease and risk for cognitive decline.
- Mice fed a high-salt diet have learning and memory problems.
- They accumulate phosphorylated and insoluble tau in the brain.
- Strains lacking tau genes, or treated with anti-tau antibodies, are spared.
Admittedly, at eight to16 times the norm, the amount of salt the mice consumed exceeds all but the very highest equivalents in which people might indulge. Still, researchers found the results thought-provoking. “However artificial the diet, this highlights that salt has effects independent of high blood pressure and that salt is a risk factor in its own right,” said Joanna Wardlaw, University of Edinburgh. Wardlaw thinks the mechanism may explain some clinical observations. “We’ve seen in studies of small stroke that despite treating high blood pressure, people continue to get worse clinically and on their brain scans,” she told Alzforum. “We need to think about the role of other common risk factors, including dietary salt.”
Li-Huei Tsai and Joel Blanchard, Massachusetts Institute of Technology, found the Weill Cornell group’s work fascinating. “They illustrate that neuronal cells and the cerebrovasculature have dynamic molecular and biochemical interactions that clearly influence neurodegenerative pathologies,” they wrote to Alzforum (full comment below). Faraco found the salt-induced reduction in nitric oxide (NO) boosted levels of p25, which activates the kinase Cdk5. Tsai has linked p25/Cdk5 to neurodegeneration (Dec 1999 news).
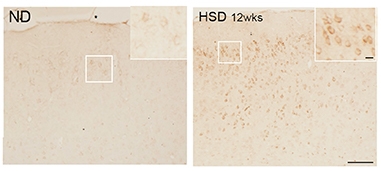
Pickled. AT8 immunostaining detects phosphorylated tau in the brains of mice fed a high-salt diet (right), but not in brains of mice on normal chow (left). [Courtesy of Giuseppe Faraco et al., Nature.]
The NO link most intrigued Zvonimir Katusic, Mayo Clinic, Rochester, Minnesota, as well. Susan Austin in Katusic’s lab found that knocking out endothelial nitric oxide synthase (eNOS) increases processing of Aβ precursor protein and impairs learning and memory, and most recently that it boosts p25 and phosphorylation of tau (Austin et al., 2010; Austin et al., 2013; Katusic and Austin et al., 2016). In Chicago, Austin reported that microglia from eNOS knockouts ramp up production of ADAM17, the primary sheddase for TREM2, and tone down production of the anti-inflammatory cytokine IL-10. “It appears release of NO by the endothelium is an important control mechanism for the brain,” said Katusic.
The plot gets thicker. The effect of high salt may not start in the endothelial cells of the brain, but in immune cells of the gut. Last year Faraco reported that a high-salt diet elicits a flood of interleukin-17 from T helper cells in the intestine. That IL-17 lead to a dearth of endothelial NO and impaired memory (Jan 2018 news). The IL-17 reduced cerebral blood flow by about 25 percent, but Faraco considers this insufficient to cause the memory impairment. Since tau pathology has been linked to cerebrovascular disease, he decided to see if a high-salt diet affected the microtubule binding protein.
Faraco put normal C56/Bl6 mice on a diet comprising 8 percent NaCl. This is 16 times the normal amount of salt in mouse chow; seawater is about 3.5 percent NaCl. The mice ate as much food as usual, but over the next 36 weeks, levels of phosphorylated tau rose. AT8 immunoreactivity peaked after 24 weeks, RZ3 immunoreactivity after 36 weeks. These antibodies recognize tau phosphorylation at serine 202/threonine 205 and threonine 231, respectively. Hyperphosphorylation of tau was detected in both male and female mice, and in mice on a 4 percent NaCl diet, albeit only AT8 staining in that case. Faraco found similar tau changes when he fed 8 percent salt to Tg2576 mice, which model amyloidosis. Levels of Aβ were unaffected.
What about neurofibrillary tangles? Faraco found none in any of the mice, but levels of insoluble tau released by formic acid did increase slightly in the cortices and hippocampi of mice on the high-salt diet.
In parallel with the tau phosphorylation, C57/Bl6 mice began having learning and memory problems. They struggled to recognize novel objects in their cages and had trouble finding the escape route in the Barnes maze. The deficits modestly correlated with AT8 binding in the cortex and hippocampus.
Was hyperphosphorylation of tau to blame? The authors tested this in two ways. They administered anti-tau antibodies to wild-type mice on high salt, and they fed high salt to tau knockouts. In both cases the animals performed as well as mice on normal chow, despite hypoperfusion of the brain, suggesting that indeed it was the tau that drove the cognitive decline due to the salt and not reduced blood flow.
Given Katusic’s prior data suggesting links between endothelial NO and tau phosphorylation, Faraco tested if he could stop the protein modification with L-arginine, a precursor in NO production. This suppressed both tau phosphorylation and the learning and memory deficits. In addition, elevated p-tau in eNOS knockouts could not be boosted further by high salt, supporting the idea that suppression of endothelial NO was behind the tau modification.
Delving more deeply into the mechanism, Faraco found that the salty food elevated calpain activity in the brain. Calpain cleaves p35 to p25; in keeping with this, the levels of the smaller peptide rose, as did activity of Cdk5, the tau kinase. All told, the data suggest that by triggering IL-17 production in the gut, high salt triggers loss of endothelial NO, which in turn leads to phosphorylation of tau and cognitive impairment.
Precisely how NO is suppressed remains to be seen. Katusic emphasized that the gas easily diffuses. Since cells in the brain are rarely more than 15 micrometers away from a blood vessel, NO could be an important signaling molecule. Faraco found no gross changes in astrocytes, microglia, or neurons in mice on high salt, as judged by GFAP, Iba1, and NeuN staining, but agreed it would be important to study downstream effects on these cells.
In her SfN talk, Austin reported that NO affected microglia more profoundly. In cultures of the cells from eNOS knockout mice, she found not only an increase in ADAM17, but also a decrease in cell surface TREM2. Mutations in this microglial receptor increase risk for Alzheimer’s and frontotemporal dementia. The sensor plays a central role in microglial homeostasis (Nov 2012 news; Oct 2012 news; Aug 2019 news). Austin also found that eNOS-/- microglia, either cultured or isolated from brain by cell sorting, make less TNFα and IL-10, pro- and anti-inflammatory cytokines, respectively, while at the same time ramping up phospholipase A2, which mobilizes arachidonic acid, a precursor for inflammatory molecules.
“We are slowly developing this concept that vascular mechanisms independent of perfusion affect cognitive impairment,” said Katusic. Tsai and Blanchard agreed. “Further unraveling these mechanisms will undoubtedly be a promising endeavor that will strengthen our understanding of how dietary habits influence susceptibility to age-related cognitive decline,” they wrote.
For his part, Faraco is using RNA-Seq to study what happens in the endothelial cells to reduce NO. “It will be interesting to examine interactions with other genetic and dietary risk factors, such as high-fructose or high-fat diets,” he said. He thinks it will be important to identify the tau species responsible for the effects on cognition. “We need to go much more deeply into the mechanism of neuronal dysfunction.”—Tom Fagan
References
Alzpedia Citations
News Citations
- Enzyme Essential to Brain Development Found to Hyperphosphorylate Tau, Kill Neurons
- Gut Immune Cells, not Blood Pressure, Blamed for Salt’s Effect on Brain
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
- Mutations in TREM2 Cause Frontotemporal Dementia
- In Alzheimer’s, More TREM2 Is Good for You
Research Models Citations
Paper Citations
- Austin SA, Santhanam AV, Katusic ZS. Endothelial nitric oxide modulates expression and processing of amyloid precursor protein. Circ Res. 2010 Dec 10;107(12):1498-502. PubMed.
- Austin SA, Santhanam AV, Hinton DJ, Choi DS, Katusic ZS. Endothelial nitric oxide deficiency promotes Alzheimer's disease pathology. J Neurochem. 2013 Jun 8; PubMed.
- Katusic ZS, Austin SA. Neurovascular Protective Function of Endothelial Nitric Oxide - Recent Advances. Circ J. 2016 Jun 24;80(7):1499-503. Epub 2016 May 25 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Faraco G, Hochrainer K, Segarra SG, Schaeffer S, Santisteban MM, Menon A, Jiang H, Holtzman DM, Anrather J, Iadecola C. Dietary salt promotes cognitive impairment through tau phosphorylation. Nature. 2019 Oct;574(7780):686-690. Epub 2019 Oct 23 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Picower Institute of MIT
Icahn School of Medicine at Mt. Sinai
High-salt diets have long been associated with increased risk for dementia. However, the underlying mechanisms have largely been unknown. Faraco and colleagues (Faraco et al., 2018) recently presented an intriguing mechanism in mice, showing that a high-salt diet alters the gut biome, increasing circulating IL-17, which impairs endothelial function leading to reduced cerebral blood flow. From this it is reasonable to postulate that reduced cerebral blood flow is the primary driver of cognitive impairments associated with high-salt diet.
However, Faraco and colleagues now expand upon this fascinating mechanism showing a high-salt diet can induce cognitive impairments independent of reduced cerebral blood flow. Instead, they demonstrate that a high-salt diet reduces endothelial nitric oxide, which activates neuronal Cdk5 and leads to hyperphosphorylation of tau and cognitive impairments. In humans, it remains unclear whether a high-salt diet contributes to age-related cognitive decline. However, these observations and mechanisms from mice will clearly help elucidate that relationship. Furthermore, Faraco and colleagues highlight that the importance of the brain’s microvasculature extends beyond regulating cerebral blood. They illustrate that neuronal cells and the cerebrovasculature have dynamic molecular and biochemical interactions that clearly influence neurodegenerative pathologies. Further unraveling these mechanisms will undoubtedly be a promising endeavor that will strengthen our understanding of how dietary habits influence susceptibility to age-related cognitive decline.
References:
Faraco G, Brea D, Garcia-Bonilla L, Wang G, Racchumi G, Chang H, Buendia I, Santisteban MM, Segarra SG, Koizumi K, Sugiyama Y, Murphy M, Voss H, Anrather J, Iadecola C. Dietary salt promotes neurovascular and cognitive dysfunction through a gut-initiated TH17 response. Nat Neurosci. 2018 Feb;21(2):240-249. Epub 2018 Jan 15 PubMed.
Make a Comment
To make a comment you must login or register.