Gene and Stem Cell Therapies Make Strong Showing at ALS/MND Meeting
Quick Links
More than 800 researchers, clinicians, and patients with ALS gathered in balmy Orlando, Florida, on December 11-13, for the 26th International Symposium on ALS/MND. Attendees felt a sense of urgency, but also hope—urgency because “time is not on our side,” pointed out Sally Light, chief executive of the Motor Neurone Disease Association in the United Kingdom, which organized the conference. She noted that in the United Kingdom alone, six people die of ALS every day. Yet thanks to the advances discussed at the meeting, including novel therapeutic approaches, identification of new disease genes, and a growing understanding of how pathogenic proteins move from cell to cell, attendees felt the field was making progress. “I am wildly more optimistic than I was 10 years ago,” commented Kevin Talbot of Oxford University in the United Kingdom, chair of the program committee.
That optimism was most evident at a gene therapy session, when Brian Kaspar of Nationwide Children’s Hospital in Columbus, Ohio, presented early results from a clinical trial. He treated babies with the most severe version of the motor neuron disease spinal muscular atrophy (SMA), which normally causes paralysis and death within two years of birth. Yet all of the babies in Kaspar’s trial—started in spring of 2014—remain alive and wriggling. He played videos of a baby girl, six months out from her treatment, who sat up straight without support, something untreated babies with SMA never achieve.
“These are early, Phase 1/2 clinical trials,” Kaspar cautioned. “We are certainly not claiming victory, but it is exciting.”
“I thought the preliminary Phase 1 data Kaspar showed looked very encouraging,” commented Adrian Krainer of Cold Spring Harbor Laboratory in New York, who did not participate in the work. “Larger, [placebo] controlled trials will be necessary later on to determine if there are statistically significant improvements, but it is nice to see that there have not been safety issues.” During the same session, Krainer presented an antisense oligonucleotide therapy for SMA, which has also benefited participants in ongoing trials. In a separate session, Clive Svendsen of the Cedars-Sinai Medical Center in Los Angeles described his work to link up gene therapy with another promising modern technique, stem cell treatment, for ALS. His trial starts in early 2016.
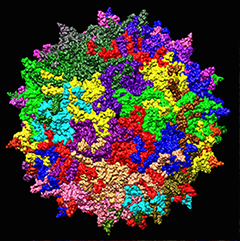
Delivery vehicle.
Scientists use adeno-associated viruses to carry genes into patients.
Gene Therapy for SMA
SMA, an autosomal recessive disease, results from missense or deletion mutations in the SMN1 gene, which codes for the survival of motor neuron protein. SMN processes mRNAs, and for unknown reasons motor neurons are particularly vulnerable when they contain two bad copies of the SMN1 gene. Once motor neurons die, muscles wither without stimulation, causing the atrophy for which the disease is named. People possess a second SMN gene, SMN2, but a single nucleotide difference from the SMN1 code leads predominantly to an alternative spliced form that lacks exon 7. This shorter isoform is unstable and rapidly degraded. Nonetheless, the little bit of full-length SMN protein produced by SMN2 can be enough to allow people with two mutated copies of SMN1 to survive. In fact, the disease has a spectrum of severity, based on how much SMN2 is expressed. Some SMN1 mutation carriers with the mildest version, type IV, show no symptoms until their 30s, but in its most severe and most common form, type I—with almost no backup SMN2 expression—the disease cripples infants.
For the clinical trial, Kaspar and colleagues employed the harmless adeno-associated virus AAV9 to deliver a pristine SMN1 gene to motor neurons. Different AAV types, with different capsid structure, target different cell types, and AAV9 is able to cross the blood-brain barrier and bind to cellular proteins that have galactose attached, attracting it to both motor neurons and astrocytes (Foust et al., 2009; Shen et al., 2011). While AAV9 preferentially transduces astrocytes in adult mice, in larger animals it targets motor neurons and oligodendrocytes as well, Kaspar said.
Once it finds its target cells, the virus slips its genetic material into the nucleus, where it remains as an extrachromosomal bit of DNA. That DNA persists, so theoretically one treatment should provide SMN protein for a lifetime. In a mouse model of severe SMA, this treatment preserved mobility and mice survived past 27 weeks, whereas untreated animals survived around two weeks (see Mar 2010 news).
Kaspar licensed the treatment to the gene therapy company AveXis Inc., which is collaborating with Nationwide Children’s on a trial sited at the hospital. The study will enroll up to 18 infants, nine months of age or younger. All participants must have SMA due to SMN1 mutations on both chromosomes, with symptoms starting before six months of age, and low muscle tone with delayed motor skills. They will receive the one-time treatment with AAV9 carrying the SMN1 gene as an intravenous infusion, over about an hour. All participants receive treatment; there is no placebo arm. However, the study will test three doses of the virus, starting low and increasing in later enrollees. The authors suspect higher doses may work better, perhaps by transducing more motor neurons, or possibly by increasing SMN1 expression in individual cells or infecting glia as well as motor neurons, said collaborator Kevin Foust of Ohio State University in Columbus.
At the lowest dose, three babies received 67 trillion virus particles per kilogram body weight. The scientists have FDA approval to try 200 trillion per kilogram in up to nine more babies, and the highest titer of 330 trillion in the final participants. The researchers mainly will be looking for any toxic effects of the treatment, but will also assess the age at which babies either die or require a ventilator to breathe. They will compare their results to the standard progression of severe SMA (Finkel et al, 2014). SMA studies often rely on death or the requirement of a ventilator to breathe as a combined endpoint. Ordinarily, half of these babies would die or require a ventilator to breathe by 10.5 months, Kaspar said, and 92 percent would die or require ventilation by 20 months. In addition, the trial assesses movement by a standard score, with items such as gripping the tester’s hand while being lifted into a sitting position (Glanzman et al., 2010). The trial is expected to finish in June of 2017.
In Orlando, Kaspar reviewed the data so far. Three patients have received the lowest dose of the virus, and seven more received the intermediate dose of about 200 trillion per kilogram. The researchers have not tried the highest dose yet. Babies tolerate the infusion while awake with no problems, Kaspar said. From April of 2014, when the first baby was enrolled, to September 30, the last point at which the data were logged, Kaspar said that no babies in the study have died or required ventilation. They also moved better than they did before gene therapy, and the ones who received the intermediate dose benefited more than those who received the low dose. Though infants with severe SMA never manage to sit up on their own, the girl in Kaspar’s video could. The trial will continue with up to eight more babies, including some at the highest viral dose.
SMN Backup Therapy
AAV gene therapy can deliver a working SMN1 gene, but what if people could make use of the SMN2 gene they already have? It shares 99 percent of the SMN1 nucleotide sequence. In coding regions, SMN2 differs from SMN1 by a single cytosine-to-thymine switch, which prevents exon 7 inclusion in most transcripts but does not change the amino acid code. Krainer sought to promote exon 7 inclusion to create full-length SMN2 protein. Because the thymine substitution reduces affinity for a splicing activator needed to retain exon 7 in the mRNA, Krainer thought if he blocked a splicing repressor that sits nearby, he might coax the splicesome to incorporate exon 7.
To do this, Krainer collaborated with Isis Pharmaceuticals of Carlsbad, California, to make antisense oligonucleotides (ASOs) specific for the repressor binding site. Normally, ASOs instigate destruction of their target mRNAs by RNase H. In this case, the scientists modified the oligonucleotide backbone so RNase H does not recognize it; it just sits inert on the mRNA. Simply by being there, the ASO blocks the splicing repressor. When the spliceosome processes SMN2 hnRNA, now missing both a splicing activator and a repressor, it should incorporate exon 7, the theory goes. In fact, in SMA model mice, the ASO bumps up exon 7 inclusion from 15 percent to more than 80 percent, and extends lifespan from 10 days to an average of eight months at the maximum dose the researchers have tried (see Mar 2011 news; Hua et al., 2010).
The treatment, first called ISIS-SMNRx and rechristened Nusinersen, is now wending its way through clinical testing, Kainer said. Because oligonucleotides usually do not cross the blood-brain barrier, physicians infuse the drug via lumbar puncture. The oligonucleotide disappears from cerebrospinal fluid with a half-life of four to six months, so the doctors infuse it at regular intervals—every six months in children, and every four months in infants since their disease is more severe. In 2014 Isis announced that in two open-label Phase 2 trials, one of infants and one of older children, the ASO improved movement scores and survival. At that time, 11 of 16 infants who had received three doses of Nusinersen were still alive, with an average age at death or ventilation of 16.3 months for those on a 6 mg dose and 13.8 months for those on a 12 mg dose, compared to 10.5 months in the natural history study of untreated babies. For 14 of the participants, their motor scores also rose compared to baseline assessments, with an average increase of 9.3 points out of a possible maximum score of 64. As Krainer reported in Orlando, a couple of those babies who died of pneumonia came to autopsy, and scientists observed more SMN RNA and protein in their nervous system tissues.
In a trial of older children with SMA type II or III, aged two to 12 years old, Isis provided multiple doses of 1-12 milligrams of Nusinersen to 56 children, it reported in 2014. These kids also got stronger, improving by 1.5 to 3.7 points, depending on dose, out of a maximum of 66 on an expanded version of the Hammersmith Functional Motor Scale. In Orlando, Krainer said that 30 children continue with the treatment on an open-label extension, so some have been receiving the therapy for nearly four years.
Now, the company is recruiting for two Phase 3 studies, one of 111 infants aged up to seven months, and one of 117 children, aged two-12 who have a later-onset form of the disease. In each study, two-thirds of participants will receive Nusinersen, while one-third will receive sham treatment. The studies, started in 2014, are expected to wrap up in mid 2017.
Researchers at the meeting were thrilled by the progress in treating SMA, and said it gave them hope for treating ALS. “SMA is a trailblazer here,” commented Talbot, who did not participate in either trial. “It is showing that genetic disorders are, in a sense, tractable.” Similar treatments for familial ALS are already in preclinical and early clinical testing (see also Nov 2014 conference news; Jun 2015 news; May 2013 news).
ALS to Follow?
Kaspar and colleagues have made progress in developing therapies to shut down expression of the mutant SOD1 gene, which is responsible for about 10 percent of familial ALS. He used AAV9 to deliver short, hairpin RNAs that silence SOD1 to the spinal cords of transgenic mice that model the disease. In mice carrying human SOD1-G93A, which normally die at around 19 weeks, a one-time treatment improved survival by up to seven weeks. In a milder model that expresses SOD1-G37R, the treatment extended the 13-month average survival by three months (see Sep 2013 news).
The AAV9 virus included a gene for green fluorescent protein to mark infected cells, making it unsuitable for use in people. Kaspar and collaborators are now testing a new version without GFP. In the severe mouse model, some animals have lived beyond six months after a single treatment delivered when they were a day old. “To see this G93A mouse live beyond 200 days is really shifting the curve on survival,” Kaspar said. If the treatment proves safe in mice and macaque monkeys, he hopes to move forward with clinical studies.
Talbot said researchers still have some work to do to perfect this approach, pointing out that in the animal models SOD1 expression dropped, but not down to zero. “If you have a small amount of misfolded SOD1 in the aging nervous system, I think that could trigger disease,” he said. Foust suggested that the treatment would probably not eliminate disease completely, but over time would reduce SOD1 aggregation and slow or halt its course.

Perfecting Transfection.
Scientists used AAV9 to deliver SOD1 antisense oligonucleotides and green fluorescent protein (brown) to the spinal cord of a mouse. [Courtesy of Sara Gombash Lampe, Ohio State University, Columbus.]
In the closing session of the meeting, Svendsen explained how he combines gene and stem cell therapies in one. He noted that though motor neurons can be created from induced pluripotent cells and transplanted into the spinal cord, those cells do not usually integrate into the neuromuscular networks or connect to muscles. Plus, they would likely be subject to the same degeneration that plagued the original motor neurons in the cord. Instead, Svendsen chose to transplant astrocytes derived from stem cells that have been genetically engineered to pump out glial-derived neurotrphic factor (GDNF) (Suzuki and Svendsen, 2008). GDNF promotes motor neuron and corticospinal neuron survival, but the protein cannot easily be delivered to the brain, Svendsen said. The transplanted cells give it a ride (Henderson et al., 1994).
In rats expressing SOD1-G93A, the transplanted astrocytes survived for up to 11 weeks and released GDNF (Klein et al., 2005). Motor neurons survived for longer, and while the rats still developed paralysis, the nerves responsible for breathing—whose demise usually causes death in people with ALS—worked better than in control animals (Nichols et al., 2013).
Svendsen plans to enroll 18 people with ALS in a Phase 1/2 trial. The trial will start with transplants to the lower spinal cord, but Svendsen suspects multiple implants along the cord may be necessary. It is hard to predict outcomes for people based on the rat work, Svendsen told Alzforum, because the rats develop a severe disease due to multiple copies of the SOD1 transgene. The transplants might only improve breathing, or they might reduce paralysis too, he speculated.
Neurosurgeons will choose one side of the spinal cord to implant, based on where they can more easily avoid blood vessels that zigzag across the cord, Svendsen said. Since the stem cells do not cross from one side of the spinal cord to the other, the unimplanted side will serve as a control. Only the surgeon will know which side received the astrocytes, leaving everyone else involved in the study blinded. ALS symptoms typically progress at similar rates in both legs, Svendsen said, so the researchers will be hoping for slowed progression on one side, using a chair-like device that measures muscle strength to monitor symptom spread (see Oct 2011 news). “If something is happening, we should be able to detect it,” Svendsen said. He told Alzforum that if the scientists do detect benefit, they will offer participants implants in the other side of their spinal cords as well. He expects to enroll the first participant in the two-year trial in March of 2016.—Amber Dance
References
News Citations
- Baby Steps for Gene Therapy? Fatal Infant Disease Cured in Mice
- Research Brief: Researchers Solicit SMN Understudy to Treat SMA
- Listen Up, Gene Silencing Strikes a Chord at RNA Meeting
- Scientists Eager to Test ALS Gene Therapy
- Paper Alert: Antisense Oligonucleotide Therapy Safe for ALS?
- Lou Gehrig’s RNA Interference Success in Mice, Monkeys
- NEALS: Desperately Seeking ALS Biomarkers
Paper Citations
- Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009 Jan;27(1):59-65. PubMed.
- Shen S, Bryant KD, Brown SM, Randell SH, Asokan A. Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J Biol Chem. 2011 Apr 15;286(15):13532-40. Epub 2011 Feb 17 PubMed.
- Finkel RS, McDermott MP, Kaufmann P, Darras BT, Chung WK, Sproule DM, Kang PB, Foley AR, Yang ML, Martens WB, Oskoui M, Glanzman AM, Flickinger J, Montes J, Dunaway S, O'Hagen J, Quigley J, Riley S, Benton M, Ryan PA, Montgomery M, Marra J, Gooch C, De Vivo DC. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014 Aug 26;83(9):810-7. Epub 2014 Jul 30 PubMed.
- Glanzman AM, Mazzone E, Main M, Pelliccioni M, Wood J, Swoboda KJ, Scott C, Pane M, Messina S, Bertini E, Mercuri E, Finkel RS. The Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): test development and reliability. Neuromuscul Disord. 2010 Mar;20(3):155-61. Epub 2010 Jan 13 PubMed.
- Hua Y, Sahashi K, Hung G, Rigo F, Passini MA, Bennett CF, Krainer AR. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010 Aug 1;24(15):1634-44. PubMed.
- Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simmons L, Moffet B, Vandlen RA, Simpson LC corrected to Simmons L, Koliatsos VE, Rosenthal A. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science. 1994 Nov 11;266(5187):1062-4. PubMed.
- Klein SM, Behrstock S, McHugh J, Hoffmann K, Wallace K, Suzuki M, Aebischer P, Svendsen CN. GDNF delivery using human neural progenitor cells in a rat model of ALS. Hum Gene Ther. 2005 Apr;16(4):509-21. PubMed.
- Nichols NL, Gowing G, Satriotomo I, Nashold LJ, Dale EA, Suzuki M, Avalos P, Mulcrone PL, McHugh J, Svendsen CN, Mitchell GS. Intermittent hypoxia and stem cell implants preserve breathing capacity in a rodent model of amyotrophic lateral sclerosis. Am J Respir Crit Care Med. 2013 Mar 1;187(5):535-42. PubMed.
External Citations
Further Reading
Papers
- Azzouz M, Le T, Ralph GS, Walmsley L, Monani UR, Lee DC, Wilkes F, Mitrophanous KA, Kingsman SM, Burghes AH, Mazarakis ND. Lentivector-mediated SMN replacement in a mouse model of spinal muscular atrophy. J Clin Invest. 2004 Dec;114(12):1726-31. PubMed.
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011 Oct 6;478(7367):123-6. PubMed.
- Thomsen GM, Gowing G, Svendsen S, Svendsen CN. The past, present and future of stem cell clinical trials for ALS. Exp Neurol. 2014 Mar 6; PubMed.
- Thomsen GM, Gowing G, Latter J, Chen M, Vit JP, Staggenborg K, Avalos P, Alkaslasi M, Ferraiuolo L, Likhite S, Kaspar BK, Svendsen CN. Delayed disease onset and extended survival in the SOD1G93A rat model of amyotrophic lateral sclerosis after suppression of mutant SOD1 in the motor cortex. J Neurosci. 2014 Nov 19;34(47):15587-600. PubMed.
News
- Honolulu: So Far, So Good in Stem Cell Safety Study for ALS
- Stem Cells Pass Initial Safety Muster in ALS, Multiple Sclerosis
- Drug Data Dash at AAN Annual Conference
- Chicago—ALS Clinical Trials: New Hope After Phase 3 Setbacks
- Ready or Not: Stem Cell Therapies Poised to Enter Trials for Alzheimer’s
- Parkinson's: Update on Gene Therapy, Fetal Cell Transplants
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.