Expression, Expression, Expression—Time to Get on Board with eQTLs
Quick Links
Next-generation genetic analysis has opened the floodgates to a wave of new gene discovery. Whole-exome and -genome sequencing have nearly doubled the number of previously known Alzheimer’s genes (see Part 1 of this story). But modern geneticists have other tools available as well. One is to look for changes in gene expression in distinct brain cell populations to understand how risk variants might make people sick. “We are moving from generating genomic data to transcriptomic data,” said Elisa Navarro of the Icahn School of Medicine at Mount Sinai, New York.
- Expression studies implicate microglia in Parkinson’s, as well.
- The microglial response to amyloidosis may kick off AD pathogenesis.
- Neurodegenerative disease in general may result from a failure of protein clearance.
This approach is important, because many disease associations fall into regulatory, not coding regions. When variants affect gene expression, geneticists call them expression quantitative trait loci, and eQTLs have become a big deal in AD genetics. To find eQTLs, geneticists correlate variants with expression changes in a given cell type. eQTLs are cell-type-specific because cell types tend to contain a unique mix of different transcription factors that bind distinct enhancers. Thus, a mutation in a particular enhancer may only affect expression in one type of cell, and eQTLs therefore offer clues to the cell type most involved in the disease. For example, for Alzheimer’s disease, researchers have now linked much of the genetic risk to eQTLs in microglia (Jun 2017 news).
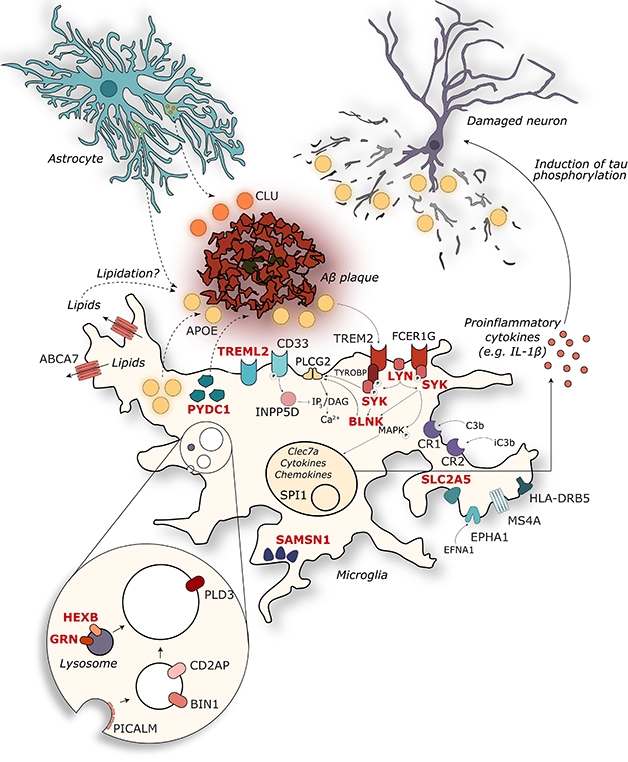
At the Heart of it All. Surprisingly many AD gene variants exert their effect in microglia, where they influence the response to amyloidosis. [Courtesy of Annerieke Sierksma.]
In Lisbon, speakers presented similar findings for Parkinson’s disease. Navarro, working with Towfique Raj at Icahn, used monocytes as a more accessible proxy for microglia. She isolated monocytes from 60 people with PD and 57 aged-matched controls, and analyzed their gene expression with RNAseq. Monocytes from patients showed numerous expression changes relative to control cells, confirming dysregulation of the immune system in this disease.
Pathway analysis flagged lysosomal and mitochondrial genes as the most altered. Most of the lysosomal genes were downregulated, and indeed, cultured monocytes from patients degraded waste only sluggishly. Notably, both these pathways contain many GWAS hits for PD. So far, the researchers have identified, in these monocytes, three eQTLs that affect expression of a known PD gene—LRRK2, GPR65, and GPNMB (Ramdhani et al., 2018). Navarro’s study is underpowered to find eQTLs, but the researchers are adding samples from another 120 people, Navarro said. Though the findings implicate immune cells in PD pathogenesis, they provide no information on how gene expression may change in the brains of people with the disease.
Regina Reynolds, working with Mina Ryten of University College London, took a different dig at the biology underlying GWAS associations. She examined whether genes linked to PD were expressed more or less highly in different brain cell types—neurons, astrocytes, oligodendrocytes, or microglia. She saw no consistent difference. Then the researchers dug for any association of PD genes with chromatin regulation in different cell types, and again came up empty. “We were getting a bit frustrated,” Reynolds said in Lisbon.
The researchers reframed the question. Perhaps instead of Parkinson’s risk being concentrated in a particular cell type, it is in particular processes that are ubiquitous to all cells, such as autophagy, lysosomal degradation, or mitochondrial function. With that, they hit pay dirt, when pathway analysis placed Parkinson’s heritability predominantly in lysosomal genes. But do lysosomes function the same in every cell type? Here the analysis turned up a difference, finding higher expression of lysosomal genes in microglia than in neurons (Reynolds et al., 2018). “We should be using glial cells in our Parkinson’s models,” Reynolds concluded.
For Alzheimer’s disease, too, evidence is converging on microglia as a key factor (see image above). In Lisbon, Annerieke Sierksma and Ashley Lu of KU Leuven, Belgium, presented their analysis of how AD risk genes change expression in the presence of amyloid or tau pathology. Working with Bart De Strooper and Mark Fiers, the researchers compared gene expression in APPswe/PS1L166P and THY-Tau22 mice. In the latter, only a handful of AD genes changed expression. These were mostly involved in neuronal function; they were suppressed, and were suppressed stably over the life of the mouse.
By contrast, in the amyloid mice, the researchers found a massive increase in AD gene expression as amyloidosis advanced. Most of these genes clustered in the inflammatory pathway and were expressed in microglia. In addition to known genes, the analysis highlighted 11 new genes of interest: GPC2, TREML2, SYK, GRN, SLC2A5, SAMSN1, PYDC1, HEXB, RRBP1, LYN, and BLNK. All are expressed in microglia, controlled by the master regulator PU.1. All rev up in the presence of amyloid, but have not been previously linked to AD. “AD risk variants determine how microglia respond to accumulating Aβ pathology,” the researchers concluded. The data underline the idea that amyloid triggers Alzheimer’s disease (Sierksma et al., 2019).
Other researchers agreed, saying privately that their own ongoing human-expression studies indicate extraordinarily broad gene-expression increases in response to amyloid deposition, and much more limited expression changes in response to tau pathology.
John Hardy of University College London agrees with this overall conclusion. “Genetic variability in the response to amyloid is the key to AD,” he told Alzforum. Hardy noted that many AD genes affect the metabolism of membrane lipids and motility of microglia. Perhaps amyloidosis starts in the cell membrane, and cells then have to respond to membrane disruption, he proposed.
In Parkinson’s disease, on the other hand, lysosomal stress seems to be the key. Genetic variants that boost α-synuclein expression are enough to overload the lysosome (Nov 2003 news; Jun 2010 news). In tauopathies, such as frontotemporal dementia and progressive supranuclear palsy, the ubiquitin proteasome is most affected. This organelle bears primary responsibility for mopping up tau, and again, high levels of tau alone are enough to trigger pathology (Rovelet-Lecrux et al., 2010). “We’ve come to the same conclusion in all three diseases: an age-dependent failure of clearance pathways,” Hardy said.
Geneticists hope these insights will lead to new therapies. “Genetics can drive discovery of drug targets,” said Gerard Schellenberg of the University of Pennsylvania. “There is a lot of biology yet to be revealed.”—Madolyn Bowman Rogers
References
News Citations
- At AD/PD Conference, New Alzheimer’s Genes Reinforce Known Pathways
- Microglial Master Regulator Tunes AD Risk Gene Expression, Age of Onset
- Synuclein and Parkinson's—It's All in the Dose
- Excess α-Synuclein Sends Synapses Sputtering
Research Models Citations
Paper Citations
- Ramdhani S, Navarro E, Udine E, Efthymiou AG, Schilder BM, Parks M, Goate A, Raj T. Tensor decomposition of stimulated monocyte and macrophage gene expression profiles identifies neurodegenerative disease-specific trans-eQTLs. PLoS Genet. 2020 Feb;16(2):e1008549. Epub 2020 Feb 3 PubMed.
- Reynolds RH, Botía J, Nalls MA, International Parkinson’s Disease Genomics Consortium (IPDGC), System Genomics of Parkinson’s Disease (SGPD), Hardy J, Gagliano Taliun SA, Ryten M. Moving beyond neurons: the role of cell type-specific gene regulation in Parkinson's disease heritability. NPJ Parkinsons Dis. 2019;5:6. Epub 2019 Apr 17 PubMed.
- Sierksma A, Lu A, Mancuso R, Fattorelli N, Thrupp N, Salta E, Zoco J, Blum D, Buée L, De Strooper B, Fiers M. Novel Alzheimer risk genes determine the microglia response to amyloid-β but not to TAU pathology. EMBO Mol Med. 2020 Mar 6;12(3):e10606. Epub 2020 Jan 17 PubMed.
- Rovelet-Lecrux A, Hannequin D, Guillin O, Legallic S, Jurici S, Wallon D, Frebourg T, Campion D. Frontotemporal dementia phenotype associated with MAPT gene duplication. J Alzheimers Dis. 2010;21(3):897-902. PubMed.
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.