As DIAN’s Scope Widens, Funding Narrows
Quick Links
The Dominantly Inherited Alzheimer Network is going full tilt. It is collecting data on its cohort of people at risk of autosomal-dominant AD while beginning to treat some participants in a first therapeutic trial. Alas, just as the past five years of arduous groundwork are paying off in the form of longitudinal results and new scientific ideas, public funding is becoming so scarce that the DIAN researchers are increasingly asking industry to help move the project forward. DIAN’s Pharma Consortium includes 10 pharmaceutical companies that have contracted to jointly support the development of clinical trials for people with autosomal-dominant AD, regardless of whether their own drug is currently being studied in a given DIAN trial. At the most recent of the consortium’s regular meetings, on 14 April 2013 in Washington, DC, its scientists heard not only the latest scientific update on the trial and CSF biomarker challenges; they also fielded a volley of informal pitches for research opportunities that present themselves now that the observational study cohort is in place, but for which there are no funds.
“The interest in DIAN among the scientific community and families worldwide has grown, but I am concerned that we may miss opportunities at several levels—scientific, power, collaborative—because of funding,” Randy Bateman of Washington University, St. Louis, Missouri, told Alzforum. Here is a brief summary of the meeting, which drew scientists from Genentech, Genzyme/Sanofi, Janssen Alzheimer Immunotherapy, Lilly, Pfizer, Roche, the Alzheimer’s Association, and WashU.
First off, news on the clinical trial. Since Alzforum last reported on DIAN (see ARF related series), the first therapeutic trial has begun, with enrollment standing at eight people at this time. For the time being, only the WashU site is enrolling, with other sites in the startup phase. Bateman said that is to ensure all systems in this complex trial—which will eventually compare three drugs and uses an extensive suite of biomarker outcomes—are working properly before bringing online other sites and countries.
While this first trial is ramping up, DIAN scientists are already planning to change the current design of its end phase in an attempt to shave two years off the total time it would take to get a drug approved. As is, the trial would measure outcomes once people have been on drug for two years and, after that, pool all its participants into a subsequent but separate four-year trial of the most promising drug that would measure cognitive outcomes. Counting the time it takes to enroll both phases and to analyze both datasets, the total development time, even if one of the first three drugs performed well, would add up to nearly a decade.
Since the early planning phase of DIAN, Michael Krams of Johnson and Johnson led some advisors in advocating for adaptive designs. DIAN did not start out this way, because at the time it was too uncertain how it could be done, and the DIAN scientists wanted to get a trial going quickly. “In the meantime, we explored this topic and now have a good idea what parameters to adapt on,” said Bateman. Adaptive trial designs represent a new trend in the Alzheimer’s field.
The new design essentially eliminates the break between the current biomarker trial and the cognitive registration trial, and speeds enrollment for Phase 3. In order to obtain biomarker data on every participant in a trial that takes up to two years to fully enroll, the whole operation would have to effectively pause until those data are in for the last enrollee before analyzing and starting the cognitive endpoint trial. Instead, the new design would operate seamlessly as a Phase 2/3. It would conduct an interim analysis when half of the biomarker trial participants have received drug for one year, and from then on continue to enroll for the drug that shows most promise at that point. At the DC meeting, the scientists debated the details of this design. They agreed to rely on biomarker responses for their choice of which drug to take forward into Phase 3; however, for calculating how large that Phase 3 portion of the combined trial needs to be, the standard variation of the clinical measures at the interim timepoint might prove useful as well. Taking this variation into account would help DIAN adjust the sample size for Phase 3 to ensure it is adequately powered to serve as a registration trial, suggested Owen Hagino from Sanofi-Aventis. The relationship between a biomarker response to drug and the final outcome that matters to the patient—memory, thinking, and function—is still unclear.
This trial is but the first in several Bateman wants to offer families with autosomal-dominant AD. “We do not know that these first three drugs will cure the disease. Next-generation therapies are being designed to have a larger pharmacodynamic effect on their target and be safer. We want to be ready for those,” Bateman said. Toward that end, Bateman called on the 10 companies in the consortium to update the DIAN scientists who evaluate nominations with new data on their drugs, or to nominate new drugs for future trials.
Who’s That, Lurking in the Details? Definitely, It’s the Devil!
Second, what’s new on the science? DIAN scientists are beginning to analyze longitudinal data from when participants came for return visits. Their initial impression is that ostensibly small technical details of how both imaging and cerebrospinal fluid tests are processed can sway the final conclusion on important research questions.
For example, in the field of amyloid imaging, scientists are debating the pros and cons of a procedure called partial volume correction. In essence, scientists process the PET scans to compensate for patients' cortical shrinkage as their Alzheimer’s disease advances. Gray matter atrophy can create the illusion of less amyloid, when there is simply less brain left. On the other hand, the corrective maneuver itself adds a certain amount of error, and scientists have no consensus on its value. For longitudinal studies, the issue is clear that correcting for volume loss has a profound impact, said Bateman, who presented for WashU’s amyloid PET expert Tammie Benzinger. Correcting for volume loss is not only valuable, but it also completely changes the conclusion. DIAN’s initial one- to two-year longitudinal PIB scans show that, without the correction, it looks as if amyloid deposition declines once symptoms start. In contrast, when taking into account the brain volume these people lost during the time between their first and second scans, the result flipped. Analyzed this way, amyloid deposition continued to grow in some people and stayed the same in others. “There is dramatic atrophy once people are symptomatic. The concern is that the shrinking volume introduces an artifact,” Bateman said. “We recommend comparing and reporting amyloid values with and without volume correction.”
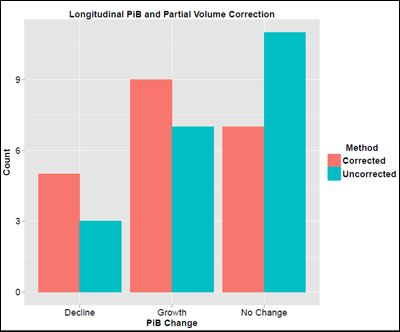
For Longitudinal Studies, Technical Fine Points Matter
Correcting for ongoing atrophy changes the interpretation of whether DIAN participants have a decline versus an increase in their brain amyloid deposition during their time in the study. Without the correction (teal), more participants had no change over the interval studied than had increases or decreases. With the correction (red), the balance shifts toward growth. Image courtesy of Tyler Blazey and Tammie Benzinger, WashU
Whether amyloid deposition goes up or down in the symptomatic phase of AD, and at what rate, is of more than passing interest. The answer to this question determines whether amyloid deposition correlates with the clinical progression of the disease, and informs decisions about which disease stage to target with amyloid-reducing drugs. It’s not clear yet whether longitudinal brain atrophy is similar in autosomal-dominant and late-onset AD (see ARF AD/PD story on longitudinal PIB data in LOAD).
Because atrophy greatly affects longitudinal amyloid PET results, Benzinger and other imaging groups have worked out a standard whereby DIAN will use the SUVR measure normalized to brainstem with a multicompartment volume correction. The first DIAN trial will use extensive brain imaging, with PIB/florbetapir comparisons, FDG-PET, and five different types of MRI. The trial data will be gathered in such a way that they are consistent with data from the DIAN observational cohort, as well as ADNI, API, and A4.
For cerebrospinal fluid tests, the situation is similar in that seemingly minute details can have an outsize effect on longitudinal measurements. Discussing first experiences with longitudinal CSF data, Anne Fagan of WashU told the consortium that variations between individual lots of the commercially available CSF Aβ and tau ELISA kits can make a big difference. This is important for a clinical trial, which is by definition a longitudinal study. For example, a given baseline sample will have a different value when analyzed right after it was drawn than when re-analyzed a year later on a different assay plate. Fagan’s group has extensively studied the reasons for variability in CSF testing. She recommended that baseline and subsequent samples from the same person always be analyzed together on the same plate and in the same test run, not years apart on separate plates. The current DIAN trial will run all baseline and one-year samples together at the time of the interim analysis. It will re-run those samples together with end-of-study samples when the trial concludes. Ideally, both runs should be done using plates from the same assay lot. Alas, the shelf life of these ELISA plates expires within a year; hence, multiple lots will have to be used in the trial.
Longitudinal data from the observational DIAN study illustrate how important these technicalities are. In this case, rerunning baseline tests upended the conclusion. First, Fagan showed cross-sectional baseline data from DIAN—that is, a one-time measurement from people of different ages. The newest dataset, now from 242 participants, confirms the result published last year from half as many people (Bateman et al., 2012). That is, older mutation carriers who are close to or past their estimated age at onset have lower CSF Aβ42 and higher CSF tau and phospho-tau than do younger carriers or non-carriers. Vilip-1, a neuronal calcium signaling molecule thought to indicate neurodegeneration, correlates with CSF tau. “These data give you the impression that various biomarkers are increasing and decreasing, just as many labs have reported in LOAD,” Fagan said in her talk.
But hold on—things are more complicated, and that story is part biology, part assay antics. For 37 of these 224 participants, Fagan now has longitudinal samples taken an average of 16 months later. In the first stab at analyzing them, tau shot up in most mutation carriers as expected, but phospho-tau did not, and Vilip-1 was no longer correlated with tau. “That worried me,” Fagan said. All of these data underwent DIAN's quality control, so sample handling was not at issue. But the longitudinal samples were run on a different assay plate. Because they came in later than the baseline samples, they were analyzed on a new plate from a different lot.
Analyzing the data by lot number explained some, but not all, of the discrepancy; therefore, Fagan’s group re-ran all samples on the same assay plate with the same lot number. Now, the pattern of tau and p-tau overlapped and tracked with Vilip-1 as expected. CSF Aβ42 behaved the same in both runs, suggesting the different assay lots had introduced error to one analyte—in this case, tau.
“Placing all samples from the same person on the same assay plate is a better way of looking at longitudinal data because it eliminates variation between plates and between lot numbers,” Fagan told the audience.
This isn’t only about plates and lot numbers, however. When analyzed correctly, the data suggest a different conclusion for how these CSF markers change over time than the cross-sectional spread across ages had seemed to imply. Specifically, levels of tau and phospho-tau would appear to increase only up to a carrier’s estimated age of onset. In people who were symptomatic, it went down. Vilip-1 showed this pattern, too. If this proves correct, it would suggest that tau marks a dynamic and robust phase of neuronal loss rather than increasing steadily throughout symptomatic disease, Fagan said in her talk. A definitive answer on this question will have to await more samples and more timepoints. “But as you can see, running all samples on the same plate makes a huge difference to how we interpret data,” Fagan told the group, “One of the biggest fallacies in biomarkers, and also in imaging, is assuming that cross-sectional data indicate longitudinal change. We really need true longitudinal data to understand progression.”
At this point, Fagan’s group is focusing on understanding all factors that throw off CSF analysis, and from that develop a best-practice protocol for the DIAN therapeutic trial. The practices may have to define details down to the exact type of collection tube and even the number of microliters to be filled into those tubes, because aliquot size can also affect the measurement. These assays are so finicky that investigators are asked to follow instructions to the T. “No flexibility allowed,” quipped Fagan. In the long run, if future assays outperform the currently available ones in head-to-head comparisons, studies including DIAN will switch, Fagan said.
DIAN’s Future: Who Will Pick Up the Tab?
With 330 participants enrolled and returning for repeat visits, DIAN’s original observation study is now poised to host ancillary studies that could be valuable to the field of Alzheimer’s research at large. For example, Bateman proposed that a current effort to establish fibroblast cell lines from DIAN participants be expanded into a bank of induced pluripotent stem cell lines derived from these fibroblasts that could be offered as tools to researchers outside of DIAN. iPSC lines of a few APP and presenilin mutations already exist, but they are scattered across different institutions (see ARF related news story; ARF news story; ARF news story). A comprehensive set reflecting multiple mutation-positive and -negative cell lines from each of the four different APP and 31 different presenilin mutations represented in DIAN would facilitate cell biology studies of the final common pathway by which all these slightly different mutations lead to the same disease. They could also help scientists in academia and pharma with target validation studies and with testing of candidate drugs on a human-derived model of AD.
Secondly, Bateman proposed new genetics research. DIAN supports discovery testing—that is, a family whose inheritance pattern looks autosomal dominant can receive counseling and have their DNA screened for mutations in APP and presenilin. Families without a mutation in these genes become candidates for exome sequencing to find new, highly AD-penetrant mutations. “If they have the family pattern of autosomal-dominant AD but these three genes come back negative, that is a real opportunity for identifying new genetic mutations,” said Bateman. Currently, this is the situation for 11 of the 22 families who have thus far chosen discovery genetic testing. One exome sequence costs about $1,000. A third ancillary study could make DIAN fluid samples available for studies on candidate neuroinflammatory markers for which sufficient prior evidence warrants use of this limited resource, the scientists at the DC meeting agreed. Finally, tau PET may need to be added to DIAN once a tracer is shown to be safe and effective.
Exploratory work in these areas has begun, but to expand and complete them, DIAN needs additional funds, Bateman said. An even bigger gap has opened up for the continuation of the DIAN parent grant, awarded in 2008 by the National Institute on Aging for an anticipated 240 participants. DIAN is applying for a five-year renewal this spring, but the renewal has been capped to the original 2008 funding levels. DIAN has been enrolling better than expected, however, with 330 people in the study now and 70 more likely to join by end of next year. This success creates a funding shortfall for 160 participants. “We are grateful that we can apply for renewal, but the reality in this time of sequestration is that funds will be severely restricted,” Bateman said.
“These exploratory research proposals are of great scientific interest, but the rubber hits the road when we go back to request funding from our respective companies, where spending has been ever greatly scrutinized,” Enchi Liu of Janssen AIP in South San Francisco told Alzforum. Because public-private partnerships have been successful in AD research, companies are receiving many more funding requests of this sort.
Funding holes also hold back the worldwide expansion of DIAN. For example, researchers in Barcelona have started a study using the DIAN protocol. They are covered for all components except amyloid PET scans. “If we can support that, then there would be a complete protocol in Spain,” said Bateman. A network of DIAN sites in Italy has started, but it lacks DIAN funding for integration with the DIAN database. In the absence of other funding, grant proposals for this list of needs will go to the pharma consortium.—Gabrielle Strobel.
References
News Citations
- DIAN Trial Picks Gantenerumab, Solanezumab, Maybe BACE Inhibitor
- From Natural History, A "Renaissance" for Amyloid Hypothesis
- Alzheimer’s Neurons Made to Order: Direct Conversion From Skin Cells
- Induced Neurons From AD Patients Hint at Disease Mechanisms
- iPSC Disease Models Up and Coming for AD, Down’s, ALS
Paper Citations
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012 Aug 30;367(9):795-804. PubMed.
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
Case Western Reserve University
Some of the events highlighted in Alzforum over the last two weeks underscore the challenge facing the Alzheimer’s disease research field, both in terms of research direction and policy administration. Identification of an innate immune network (Zhang et al., 2013) as a causative module in late-onset AD (LOAD) cases reinforces the critical role of inflammation in disease pathogenesis. Conspicuous by their absence in the LOAD networks are the genes involved in amyloid generation or degradation. One interpretation of this exhaustive study that involved thousands of human brain samples is that, contrary to the currently prevalent belief, amyloid is not a significant player in LOAD. This interpretation is further supported by the recent findings that Aβ peptides exert beneficial, anti-inflammatory effects in vivo (Kurnellas et al., 2013). So, by continuing to invest more resources in anti-amyloid trials, the results of which can be several years away, are we pursuing a strategy where the risk-to-reward ratio looks increasingly unfavorable?
This brings us to the policy issues facing the field—mainly of the research priorities in the environment of shrinking funding, nicely articulated in Gabrielle Strobel’s piece above. Few would argue against the merit of the DIAN studies, but the fact remains that numbers of DIAN cases are minuscule compared to the number of LOAD cases. Furthermore, the assumption that the pathogenesis of genetically inherited AD is homogeneous and identical or nearly identical to LOAD is increasingly looking shaky (e.g., neuroimaging data on DIAN vs. LOAD brains). Thus, although it is nice to see DIAN, A4, and other early prevention studies receiving financial support from the NIH, voices advocating increased support for basic research, which has steadily been losing its prominence over the past decade, do not seem to be heard as loudly. In the absence of increased support for basic research into disease pathogenesis, should the current anti-amyloid therapies fail to yield an effective treatment, then we will find ourselves in a dire predicament. We must diversify our research investments now to tilt the risk-to-reward ratio to a more favorable balance.
References:
Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013 Apr 25;153(3):707-20. PubMed.
Kurnellas MP, Adams CM, Sobel RA, Steinman L, Rothbard JB. Amyloid fibrils composed of hexameric peptides attenuate neuroinflammation. Sci Transl Med. 2013 Apr 3;5(179):179ra42. PubMed.
University of Melbourne
Sanjay Pimplikar is a regular commentator on Alzforum. In the current comments, I think the view promoted is too pessimistic. Although we are already in a dire predicament today because there are no effective therapies for AD, there is considerable hope on the horizon for a number of different strategies, including those outlined in this news piece on DIAN’s forthcoming trials. I certainly agree that there always needs to be a balance between basic and applied research.
Specific negative comments by Pimplikar on the Aβ-amyloid theory of AD are not supported by a wealth of evidence. For example, the rates of change of Aβ deposition, cerebral atrophy, and cognitive impairment for LOAD are very similar, with data coming from the DIAN study (see Villemagne et al., 2013). Reference to the paper by Zhang et al., 2013, as throwing doubt on the Aβ theory of AD is simply incorrect. The gene-expression profile of brain tissue from 376 LOAD brains compared with 173 “normal” brains showed that microglia are active in AD. Although we’ve known this for more than a century, the observation is entirely consistent with failure of Aβ clearance by a few percentage points in LOAD (Mawuenyega et al., 2010). Failure to quote Griciuc et al., 2013, in which CD33 in microglia is a risk factor involved in Aβ clearance, also places the Zhang paper out of context.
Now is not the time to give up on the only viable theory of AD etiology—only rigorous resting of the Aβ theory with specific therapeutic intervention will yield scientific progress.
References:
Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O, Szoeke C, Macaulay SL, Martins R, Maruff P, Ames D, Rowe CC, Masters CL, Australian Imaging Biomarkers and Lifestyle (AIBL) Research Group. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013 Apr;12(4):357-67. Epub 2013 Mar 8 PubMed.
Zhang B, Gaiteri C, Bodea LG, Wang Z, McElwee J, Podtelezhnikov AA, Zhang C, Xie T, Tran L, Dobrin R, Fluder E, Clurman B, Melquist S, Narayanan M, Suver C, Shah H, Mahajan M, Gillis T, Mysore J, MacDonald ME, Lamb JR, Bennett DA, Molony C, Stone DJ, Gudnason V, Myers AJ, Schadt EE, Neumann H, Zhu J, Emilsson V. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013 Apr 25;153(3):707-20. PubMed.
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010 Dec 24;330(6012):1774. PubMed.
Griciuc A, Serrano-Pozo A, Parrado AR, Lesinski AN, Asselin CN, Mullin K, Hooli B, Choi SH, Hyman BT, Tanzi RE. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013 May 22;78(4):631-43. PubMed.
Make a Comment
To make a comment you must login or register.