San Francisco: Gladstone Institute Hosts Tau Powwow
Quick Links
Never mind that it featured 29 of the world’s top tau experts reporting the latest on their favorite protein. The recent symposium at the Gladstone Institute of Neurological Disease (GIND), San Francisco, ran less like a formal meeting and more like a two and a half-day journal club. Lured by sunshine, free bagels, and a focused agenda, 150 researchers packed Gladstone’s main auditorium 28-30 March 2011 for 20 hours of presentations and powwow on some of the field’s most fundamental questions. Further illustrating growing interest in tau, the conference occurred just prior to the appearance of an in-depth review on tau by conference co-organizer Lennart Mucke and colleagues in today’s issue of Neuron (Morris et al., 2011). In San Francisco, attendees discussed how tau goes awry, what else the protein does within neurons, and exchanged the newest cellular and animal data on various therapeutic approaches about how to translate a recent surge of scientific advances into new treatments for tauopathies such as Alzheimer’s disease and frontotemporal dementias.
“Tau and Tauopathies: Pathogenic Mechanisms” was sponsored jointly by GIND and the German Center for Neurodegenerative Diseases (DZNE). Founded in 2009, this government-funded consortium applies a translational approach to basic science research. Pierluigi Nicotera, who leads the DZNE, and Eckhard Mandelkow, who, along with his wife Eva-Maria, will leave the Max Planck Institute in Hamburg to join DZNE in Bonn, teamed up with Mucke to organize the workshop. It was the first of many the research centers intend to put on annually, alternating between San Francisco and Bonn.

The Gladstone Institute for Neurological Disease, San Francisco, hosted “Tau and Tauopathies: Pathogenic Mechanisms” in March. Image credit: Chris Goodfellow
The idea for the inaugural gathering arose last summer when Mucke, who directs the GIND, Nicotera, and Mandelkow were chatting about the tau field. “There had been pretty radical discoveries that challenged longstanding dogmas,” Mucke said. Among these provocative findings was the demonstration that removing tau protects neurons (see Roberson et al., 2007; Ittner et al., 2010; Yuan et al., 2008), that neurofibrillary tangles themselves might not be so dangerous (see de Calignon et al., 2010; Sydow et al., 2011), and that tau toxicity can be regulated post-translationally not only by phosphorylation, but also by other modifications such as acetylation (Cohen et al., 2011; Min et al., 2010).
“These are far-reaching findings, some controversial, that we thought would benefit from a face-to-face discussion,” Mucke told ARF. Interactive yet focused dialogue is hard to come by at big meetings, what with attendees dashing from one room to another trying to catch talks in parallel sessions. “We wanted to create a ‘time-out scenario’ where people could huddle and think about a topic carefully for days in a row,” Mucke said. The organizers encouraged speakers to reserve the last half of their 30-minute slot for Q&A, and most did. The topic apparently hit a nerve. Even though the organizers did not start planning in earnest until four months before the event, the speakers came and the event filled to capacity in a few weeks of open registration; more than a dozen companies sent representatives.
Bruce Miller of the University of California, San Francisco, opened with a clinical perspective on tauopathies. He reviewed their major symptoms and molecular pathologies (see ARF conference story), recapped recent work by UCSF colleague William Seeley showing that brain atrophy in tauopathies maps to functional networks (Seeley et al., 2009; Zhou et al., 2010), and presented sobering data suggesting a potentially large tauopathy epidemic in military veterans with traumatic brain injury (see McKee et al., 2009; ARF related news series on TBI; and ARF Live Discussion on long-term effects of concussions in athletes). In response to questions on biomarkers and trial design, Miller said that tauopathies proceed about twice as fast clinically and on brain imaging parameters than AD. A six-month period can reveal significant changes, he noted, a point that makes tauopathies attractive for clinical trials. Furthermore, a good tau imaging agent, like Pittsburgh Compound B (PIB) is for amyloid, would help researchers detect early, even pre-symptomatic, disease, Miller said. From Seeley’s work, “there is a sense that there may be a signal 20 to 30 years before people are symptomatic.” This is true for AD as well (see ARF related news story).
How Tau Turns Toxic
Several speakers addressed the issue of tau’s pathogenicity. Tara Spires-Jones of Massachusetts General Hospital, Charlestown, laid out three lines of evidence for why tangles may not be as toxic as commonly believed. First, using multiphoton imaging to peer into the brains of tauopathy mice, her lab has shown that tangles form in neurons after caspases get activated, not before (Spires-Jones et al., 2008; de Calignon et al., 2010). Tangle-bearing cells also stuck around a while after imaging, suggesting that the tangles themselves are not what kills the neurons.
Second, tangle-laden neurons appear functional in circuits. This came out of experiments using rTg4510 mice with doxycycline-regulatable expression of mutant human tau. These animals respond poorly to environmental enrichment, as judged by their blunted upregulation of the immediate early gene Arc in the cerebral cortex. However, when a six-week diet with doxycycline turned off the tau transgene, the mice’s Arc response returned to normal. Importantly, tangle-bearing and non-tangle-bearing neurons seemed equally responsive in this way.
The third piece of evidence calling into question the toxicity of tangles comes from array tomography studies examining the effect of these tau aggregates on mitochondrial transport in the same tauopathy mice. Initial experiments showed that the distribution of mitochondria was disrupted near tangles in the brains of 5.5-month-old rTg4510 mice. “Finally, something a tangle is bad for—it’s not letting mitochondria get where they need to go,” Spires-Jones recalled thinking at the time. However, once again, after the researchers suppressed transgene expression, mitochondrial distribution was restored even near tangles. “So it seems that the presence of aggregated tau is not what is causing the mitochondrial deficit,” Spires-Jones said.
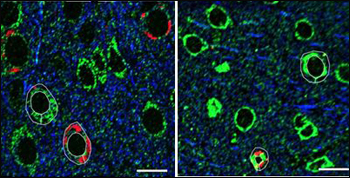
Array tomography of rTg4510 mouse brain tissue stained for mitochondria (green), aggregated tau (Alz50, red), and tubulin (blue). Left panel shows a reduction of mitochondrial area fraction in Alz50-positive cells, which is reversed in animals with six weeks of doxycycline treatment to suppress tau transgene expression (right). Scale bars 10 microns. Image credit: Kathy Kopeikina
Lester (Skip) Binder of the Feinberg School of Medicine at Northwestern University, Chicago, Illinois, also hit on the relationship between conformation and toxicity. In collaboration with Scott Brady and Gerardo Morfini at the University of Illinois at Chicago, Nick Kanaan of the Binder lab studies how misshapen tau affects axonal transport in squid axoplasm—a convenient system for cell-free assays. “The squid has two big axons,” Binder said. “You can cut them out and roll out the axoplasm.” Even with membranes removed, transport maintains its polarity, allowing researchers to study motility and biochemistry right under the microscope. In this assay, tau monomers did not affect axonal transport; however, anterograde transport drastically slowed in the presence of tau aggregates, Binder reported. The researchers pinned this effect to a conserved stretch of 17 amino acids at the N-terminus of tau (Lapointe et al., 2008). Using an antibody they generated against synthetic tau oligomers, Binder and colleagues confirmed that this N-terminal region is exposed in oligomeric tau but not filamentous forms as exist in tangles. In the May 6 Journal of Biological Chemistry, Binder’s group formally published their characterization of tau oligomers shown last year at the Society for Neuroscience conference (see ARF conference story on Patterson et al., 2011). Binder’s and Spires-Jones’s observations bolster the emerging idea that neurofibrillary tangles may not be the most damaging form of tau, after all.
But if that is true, then how does monomeric tau morph into something that wreaks havoc on synapses and, eventually, learning and memory? This puzzle fuels ongoing research at the DZNE, where researchers led by Eckhard and Eva-Maria Mandelkow are unraveling connections between tau’s structure and its toxicity. Structural analyses are challenging because tau, like about 30 percent of all proteins, is intrinsically disordered. “One consequence of this is that the protein occupies a larger space in solution,” Eckhard Mandelkow said, noting that tau sweeps out a volume 27 times that of globular proteins with comparable weight. In solution, tau’s C- and N-terminal ends fold over the repeat domain, giving the protein a “paperclip” shape (Jeganathan et al., 2006; Jeganathan et al., 2008). Moreover, the scientists have shown that the regions of tau causing it to aggregate are small relative to the whole protein, and that the stretches associating with microtubules are the same as those needed for tau’s assembly into filaments. “The physiological and pathological functions of tau are encoded in the same parts of the molecule,” Eckhard Mandelkow said.
Nuclear magnetic resonance (NMR) studies (see Mukrasch et al., 2009) are yielding some insight into tau’s secondary structure. “Its conformations are never steady. All parts of the tau chain are mobile all the time,” Eckhard Mandelkow said. “You can think of tau as a collection of freely mobile amino acids held together by some bond.” Nevertheless, the researchers have figured out how to make aggregation-prone and non-aggregating forms of tau by tweaking residues that promote or discourage formation of β-sheets—a feature the Mandelkows believe is closely tied to tau’s toxicity.
As summarized by Eva-Maria Mandelkow, their lab generated “pro-aggregant” and “anti-aggregant” inducible tau mice (ARF related news story on Mocanu et al., 2008), and showed that only the former develops tau pathology, synaptic dysfunction, and learning and memory impairment (ARF related news story on Sydow et al., 2011).
As in the Aβ field, no one has put a finger on a particular toxic species of tau just yet. Even so, the studies lend hope that targeting tau aggregation could work as a therapeutic approach. Turning off mutant tau expression in the “pro-aggregant” mice allowed them to regain their synapses and cognitive function even while tangles remained. Furthermore, aggregation-blocking compounds can counteract toxic effects of aggregation-prone tau fragments in cell models (Khlistunova et al., 2006)—and potentially in a worm tauopathy model as well.
In San Francisco, Eckhard Mandelkow played a video clip of worms expressing human tau with the P301L and V337M mutations that cause frontotemporal lobar degeneration with parkinsonism linked to chromosome 17 (FTDP-17). These worms are unusually lethargic (Kraemer et al., 2003). In collaboration with Enrico Schmidt in Freiburg, Germany, the scientists also expressed their aggregation-prone tau fragment in these worms and found it worsened their tau pathology even further, to the point of paralyzing them, Eckhard Mandelkow said. Recent studies in his lab suggest that tau aggregation inhibitors can dampen this process.
Etienne Baulieu of INSERM in Paris, France, spoke about FKBP52. This is an enzyme—a peptidyl-prolyl isomerase that may change the secondary structure of tau, presumably affecting its neurotoxicity as well. Named for its ability to bind the immunosuppressant drug FK506, FKBP52 blocks microtubule formation through its binding to tubulin (Chambraud et al., 2007). When overexpressed in PC12 rat neuronal cells, the isomerase binds tau, keeps it from accumulating, and prevents neurite outgrowth induced by nerve growth factor (ARF related news story on Chambraud et al., 2010). When overexpressed in zebrafish, it shortens growth cone processes, and the posterior lateral line nerve develops abnormally. The enzyme also worsens motor development in P301L tau transgenic fish made by Dominik Paquet, Bettina Schmid and Christian Haass at the Ludwig-Maximilians University in Munich, Germany (see ARF conference story), Baulieu reported.
By confocal imaging, Baulieu and colleagues observed FKBP52 co-localizing with tau in brain cells embryonic wild-type mice and from postmortem brain samples of normal elderly, but not in samples from people with AD or various frontotemporal dementias. Though it is not clear how the FKBP52-tau association ties in with disease, the fact that these two proteins co-localize in normal brains, and not just in cell culture systems, hints at physiological relevance and jibes with FKBP52’s hypothesized protective action on tau, Baulieu said.
Meanwhile, researchers continue grappling with tau toxicity at the level of post-translational modifications and interacting proteins. For more on these developments, read Part 2.—Esther Landhuis.
This is Part 1 of a four-part series. See also Part 2, Part 3, Part 4. Read the PDF of the entire series..
References
News Citations
- Indianapolis: Frontotemporal Dementia Research Comes of Age
- Stress and Trauma: New Frontier Lures Alzheimer’s Researchers
- Detecting Familial AD Ever Earlier: Subtle Memory Signs 15 Years Before
- Chicago: Tau and α-Synuclein Oligomers Follow Aβ Footsteps
- Tau Roundup: Inducible Mice Accentuate Aggregation and More
- Making It Stick—Tau Toxicity Linked to Aggregation Propensity
- Shall We Dance? Isomerases Twist With Tau, Synuclein, Ca2+ Channels
- Prague: Tau-Laden Neurons in Zebrafish Glow, Perish on Candid Camera
- San Francisco: Making Tau Toxic—Post-translational Changes Galore
- San Francisco: Is Tau Reduction a Good Thing?
- San Francisco: Tau—Time to Shine as Therapeutic Target?
Webinar Citations
Paper Citations
- Morris M, Maeda S, Vossel K, Mucke L. The many faces of tau. Neuron. 2011 May 12;70(3):410-26. PubMed.
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007 May 4;316(5825):750-4. PubMed.
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wölfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Götz J. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer's disease mouse models. Cell. 2010 Aug 6;142(3):387-97. Epub 2010 Jul 22 PubMed.
- Yuan A, Kumar A, Peterhoff C, Duff K, Nixon RA. Axonal transport rates in vivo are unaffected by tau deletion or overexpression in mice. J Neurosci. 2008 Feb 13;28(7):1682-7. PubMed.
- de Calignon A, Fox LM, Pitstick R, Carlson GA, Bacskai BJ, Spires-Jones TL, Hyman BT. Caspase activation precedes and leads to tangles. Nature. 2010 Apr 22;464(7292):1201-4. PubMed.
- Sydow A, Van der Jeugd A, Zheng F, Ahmed T, Balschun D, Petrova O, Drexler D, Zhou L, Rune G, Mandelkow E, D'Hooge R, Alzheimer C, Mandelkow EM. Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic Tau mutant. J Neurosci. 2011 Feb 16;31(7):2511-25. PubMed.
- Cohen TJ, Guo JL, Hurtado DE, Kwong LK, Mills IP, Trojanowski JQ, Lee VM. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat Commun. 2011;2:252. PubMed.
- Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010 Sep 23;67(6):953-66. PubMed.
- Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron. 2009 Apr 16;62(1):42-52. PubMed.
- Zhou J, Greicius MD, Gennatas ED, Growdon ME, Jang JY, Rabinovici GD, Kramer JH, Weiner M, Miller BL, Seeley WW. Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain. 2010 May;133(Pt 5):1352-67. PubMed.
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009 Jul;68(7):709-35. PubMed.
- Spires-Jones TL, de Calignon A, Matsui T, Zehr C, Pitstick R, Wu HY, Osetek JD, Jones PB, Bacskai BJ, Feany MB, Carlson GA, Ashe KH, Lewis J, Hyman BT. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci. 2008 Jan 23;28(4):862-7. PubMed.
- Lapointe NE, Morfini G, Pigino G, Gaisina IN, Kozikowski AP, Binder LI, Brady ST. The amino terminus of tau inhibits kinesin-dependent axonal transport: implications for filament toxicity. J Neurosci Res. 2009 Feb;87(2):440-51. PubMed.
- Patterson KR, Remmers C, Fu Y, Brooker S, Kanaan NM, Vana L, Ward S, Reyes JF, Philibert K, Glucksman MJ, Binder LI. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem. 2011 Jul 1;286(26):23063-76. PubMed.
- Jeganathan S, von Bergen M, Brutlach H, Steinhoff HJ, Mandelkow E. Global hairpin folding of tau in solution. Biochemistry. 2006 Feb 21;45(7):2283-93. PubMed.
- Jeganathan S, Hascher A, Chinnathambi S, Biernat J, Mandelkow EM, Mandelkow E. Proline-directed pseudo-phosphorylation at AT8 and PHF1 epitopes induces a compaction of the paperclip folding of Tau and generates a pathological (MC-1) conformation. J Biol Chem. 2008 Nov 14;283(46):32066-76. PubMed.
- Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009 Feb 17;7(2):e34. PubMed.
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schönig K, Bujard H, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008 Jan 16;28(3):737-48. PubMed.
- Khlistunova I, Biernat J, Wang Y, Pickhardt M, von Bergen M, Gazova Z, Mandelkow E, Mandelkow EM. Inducible expression of Tau repeat domain in cell models of tauopathy: aggregation is toxic to cells but can be reversed by inhibitor drugs. J Biol Chem. 2006 Jan 13;281(2):1205-14. PubMed.
- Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A. 2003 Aug 19;100(17):9980-5. PubMed.
- Chambraud B, Belabes H, Fontaine-Lenoir V, Fellous A, Baulieu EE. The immunophilin FKBP52 specifically binds to tubulin and prevents microtubule formation. FASEB J. 2007 Sep;21(11):2787-97. PubMed.
- Chambraud B, Sardin E, Giustiniani J, Dounane O, Schumacher M, Goedert M, Baulieu EE. A role for FKBP52 in Tau protein function. Proc Natl Acad Sci U S A. 2010 Feb 9;107(6):2658-63. PubMed.
Other Citations
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.