Reshuffle Parkinson’s Genetics to Lay Out Its Pathways?
Quick Links
This penultimate article in the Alzforum series on the spectrum of overlapping neurodegenerative diseases reports the call of a founding neurogeneticist, who rocks the boat from time to time, to reorganize the genetics of Parkinson’s and related diseases. In a series of provocative talks, including one at a workshop on Dementia with Lewy Bodies and Parkinson’s Disease Dementia held last March in the German city of Kassel, and again in a plenary thereafter at the 9th International conference AD/PD 2009 in Prague, John Hardy of University College London, UK, called on his audiences to cast a critical eye over the current list of proposed Parkinson’s genes and engage in some spring cleaning. Out with some genes, in with others, and the field could benefit by seeing—voila—a common metabolic pathway for Lewy body disorders, Hardy claimed.
In essence, the idea is that if three genes currently regarded as PD genes were instead set aside as genes for a mitochondrial path to neuronal death in the substantia nigra, and if other genes known to cause clinically different diseases that nonetheless come with Lewy body pathology were included, then the field might make faster progress in understanding molecular mechanisms of Lewy body diseases. That’s because scientists would have a list of genes that all impinge in some way on the metabolism of the brain glycolipid ceramide. Two papers out this month in Archives of Neurology on one such gene—glucocerebrosidase (GBA)—support this claim by placing GBA front and center as the strongest risk factor to date for sporadic PD and DLB (Mitsui et al., 2009; Clark et al., 2009; Leverenz et al., 2009; covered in Part 9 of this series).
With such a reorganization of the PARK loci, Hardy said, scientists would avoid being misled by a bewildering variety of clinical parkinsonian phenotypes. Rather than following clinical descriptions, gene sleuths could instead grab hold of the pathology as a starting point to find the underlying gene mutations and risk variants that drive pathogenesis. In a review article published this month, Hardy and colleagues recall that basing genetics on pathology has served Alzheimer disease research well (Hardy et al., 2009). Starting with Alois Alzheimer, AD was defined from the get-go by its definitive pathology of plaques and tangles. This helped geneticists find the autosomal-dominant genes, and the molecular biology of AD took off as a field when those three genes turned out to fall into the very pathway of APP metabolism that George Glenner had anticipated with his biochemical isolation of amyloid from brain. Other clinical dementias—e.g., vascular, frontotemporal—were declared different and thus did not distract the geneticists. “Just think if we had gone for genes for dementia, not for Alzheimer disease. We would be in a terrible mess,” Hardy wrote to ARF.
Parkinson disease has a different history. First, unlike Alzheimer’s 1906 paper, James Parkinson’s eponymous description, in 1817, of the shaking palsy was clinical. Second, the breakthrough of L-Dopa therapy in the 1960s rightly reinforced the importance of recognizing the clinical features because patients responded so well to this symptomatic drug, the scientists write in their review. Later it became clear that most people with PD have Lewy bodies. But so do people who suffer from other clinical diseases, and the pathology in PD generally took a back seat to the clinical perspective. While clinical classification is fine for treatment, however, it can lead molecular pathogenesis research into a thicket and delay the delineation of the multiple protein-driven pathways that give rise, either alone or in combination, to a person’s individual combination of clinical symptoms, Hardy claims.
Such a reassessment is timely now. It would cap a decade during which genetics has risen to prominence in PD research. Before that, Parkinson’s was widely considered a sporadic disease, but the initial discovery in 1997 of an a-synuclein mutation in a Greek/Italian family with early onset PD triggered a period of genetic inquiry that currently amounts to 457 genes being tracked in the PDGene database.
Here’s a current selection of the best-known PARK loci Hardy showed in Prague:
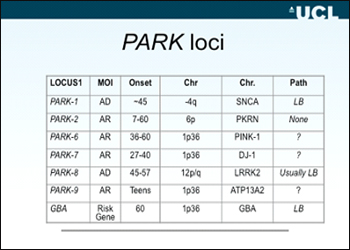
What does he find wrong with this picture? The right column shows that three well-known genes generally cause no Lewy body pathology or at least have not been reported as such yet. Parkin, Pink-1, and DJ-1 are inherited in an autosomal-recessive mode, and generally come as loss-of-function alleles. To the geneticist, proof of pathogenicity for some of those, particularly missense alleles, can be difficult to establish. Yet the primary reason Hardy suggests for grouping these three separately is that they appear to act in the same underlying pathway that is separate from the pathway of α-synuclein, LRRK2, and the new slugger in PD/DLB genetics, GBA (see part 9). Pink-1 acts upstream of parkin in the same mitochondrial pathway; DJ-1, while not directly placed in the same pathway, is an essential part of the oxidative stress response in mitochondria, too. Clinically, mutations in these three genes tend to give rise to very early onset syndromes of juvenile parkinsonism starting, in rare cases, even in childhood. In essence, Hardy suggests this: these genes are important, but they cause a mitochondrial path to parkinsonian symptoms that is probably separate from a main PD/DLB pathway that gives rise to the predominant pathology of intracellular α-synuclein aggregates. Hence, Hardy moves them to the left in his suggested pathway diagram below:

With the 3 green mitochondrial genes on the left, what’s happening on the right? Hardy suggested that the protein aggregation that leads to Lewy bodies and Lewy neurites (the main types of α-synuclein pathology visible to microcopy) encompasses a set of genes that are not typically seen as belonging to the PD spectrum, but deserve consideration. First, three smaller ones, then the big one. The little-known PLA2G6 gene causes an adult-onset parkinsonian movement disorder with cognitive and psychiatric symptoms, and the PANK2 gene causes a severe movement disorder with mental deterioration called Hallervorden-Spatz. Importantly, both of these rare diseases have Lewy pathology (Wakabayashi et al., 1999; Zhou et al., 2001; Paisan-Ruiz et al., 2008). Their proteins both also happen to map to different arms of lysosomal ceramide metabolism. When they function normally, they reduce levels of this lipid (see diagram below by Jose Bras in Hardy’s group).

Then there is the better-known Niemann-Pick Type C (NPC) gene. Recessive mutations cause the childhood-onset lysosomal storage disease known for its progressive neurodegeneration, though its phenotype varies widely. This disease might fit in because it also features Lewy bodies, and the NPC mutant from patients is known to reduce the activity of acid sphingomyelinase, an enzyme in this pathway, Hardy writes in the review. The physiological function of α-synuclein, the founding member of Lewy body disease genetics and the protein at the heart of its pathology, is still largely a mystery. But besides its synaptic localization, a biochemical role in brain lipid metabolism is likely, Hardy said (e.g., Golovko et al., 2006).
The big new anchor for this scenario, however, is GBA, the gene for the enzyme glucocerebrosidase. Five new papers published over the last 2 months alone strengthen its central role in the genetics of Lewy body diseases. Its story concludes this news series.—Gabrielle Strobel.
This is Part 8 of a nine-part series. See also Part 1, Part 2, Part 3, Part 4, Part 5, Part 6, Part 7, Part 9.
References
News Citations
- More Than Gaucher’s—GBA Throws Its Weight Around Lewy Body Disease
- Spectrum of Neurodegeneration Comes to the Fore
- Et tu, Brute? Parkinson’s GWAS Fingers Tau Next to α-Synuclein
- Neither Fish Nor Fowl—Dementia With Lewy Bodies Often Missed
- Like DLB, Like AD—Do Oligomers Stir Up the Trouble?
- Ordnung, Please—Can Biomarkers Tame a Bewildering Overlap?
- Still Early Days for α-synuclein Fluid Marker
- Meet Progranulin, The Biomarker—A Simpler Story?
Paper Citations
- Clark LN, Kartsaklis LA, Wolf Gilbert R, Dorado B, Ross BM, Kisselev S, Verbitsky M, Mejia-Santana H, Cote LJ, Andrews H, Vonsattel JP, Fahn S, Mayeux R, Honig LS, Marder K. Association of glucocerebrosidase mutations with dementia with lewy bodies. Arch Neurol. 2009 May;66(5):578-83. PubMed.
- Leverenz JB, Lopez OL, Dekosky ST. The expanding role of genetics in the lewy body diseases: the glucocerebrosidase gene. Arch Neurol. 2009 May;66(5):555-6. PubMed.
- Hardy J, Lewis P, Revesz T, Lees A, Paisan-Ruiz C. The genetics of Parkinson's syndromes: a critical review. Curr Opin Genet Dev. 2009 Jun;19(3):254-65. PubMed.
- Wakabayashi K, Yoshimoto M, Fukushima T, Koide R, Horikawa Y, Morita T, Takahashi H. Widespread occurrence of alpha-synuclein/NACP-immunoreactive neuronal inclusions in juvenile and adult-onset Hallervorden-Spatz disease with Lewy bodies. Neuropathol Appl Neurobiol. 1999 Oct;25(5):363-8. PubMed.
- Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001 Aug;28(4):345-9. PubMed.
- Paisan-Ruiz C, Bhatia KP, Li A, Hernandez D, Davis M, Wood NW, Hardy J, Houlden H, Singleton A, Schneider SA. Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol. 2009 Jan;65(1):19-23. PubMed.
- Golovko MY, Rosenberger TA, Faergeman NJ, Feddersen S, Cole NB, Pribill I, Berger J, Nussbaum RL, Murphy EJ. Acyl-CoA synthetase activity links wild-type but not mutant alpha-synuclein to brain arachidonate metabolism. Biochemistry. 2006 Jun 6;45(22):6956-66. PubMed.
Other Citations
{kind=link}
External Citations
Further Reading
News
- Spectrum of Neurodegeneration Comes to the Fore
- Et tu, Brute? Parkinson’s GWAS Fingers Tau Next to α-Synuclein
- Neither Fish Nor Fowl—Dementia With Lewy Bodies Often Missed
- Ordnung, Please—Can Biomarkers Tame a Bewildering Overlap?
- Still Early Days for α-synuclein Fluid Marker
- Meet Progranulin, The Biomarker—A Simpler Story?
- Like DLB, Like AD—Do Oligomers Stir Up the Trouble?
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.