Multiple Strategies Seek to Banish α-Synuclein Aggregates
Quick Links
For synucleinopathies, which include Parkinson’s disease, multiple system atrophy, and dementia with Lewy bodies, scientists currently have no way to slow the underlying disease progression. Many groups are on the hunt for ways to prevent or clear α-synuclein aggregates. At this year’s AD/PD meeting, held March 5 to 9 in Lisbon, Portugal, speakers presented a variety of approaches that are in early stage trials or heading that way. Some use small molecules to break up aggregates. These include an oligomer-busting drug entering Phase 2, and a new candidate that disassembles fibrils by targeting a co-aggregated synaptic protein. Other approaches use antibodies, but with a twist. One immunotherapy strategy, rather than directly targeting intracellular α-synuclein, blocks its spread by binding a cellular receptor. Another antibody binds only the nitrated form of α-synuclein, which is linked to toxicity. While it remains to be seen which approaches will pan out, the talks demonstrated that synucleinopathy researchers have disease modification squarely in their sights.
- Tomaralimab blocks internalization of α-synuclein by oligodendrocytes.
- Targeting a nitrated form of α-synuclein lowered fibril load.
- Small molecule syntacasyn splits up synapsin III/α-synuclein pairing and restores dopamine homeostasis.
- Synuclein oligomer-buster emrusolmin, aka anle138b, heads to Phase 2.
Stopping Spread by Blocking Uptake
Immunotherapy approaches against α-synuclein face a challenge—unlike in Alzheimer’s disease, where plaques are extracellular, α-synuclein deposits form inside cells. Antibodies are large molecules that do not readily cross the plasma membrane and thus may not reach aggregates. One solution is to target α-synuclein spread rather than the protein itself.
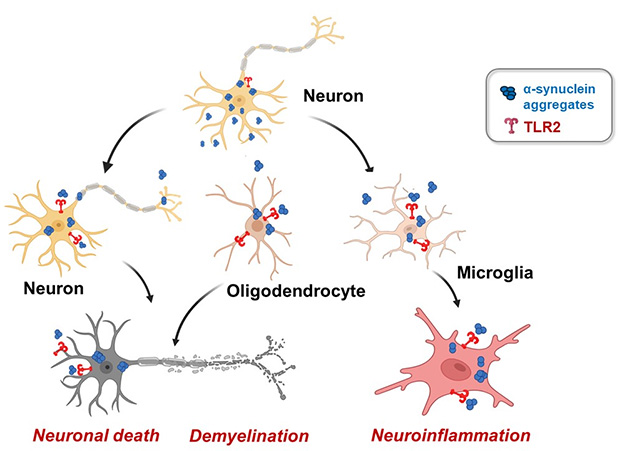
One Receptor, Multiple Actions. α-Synuclein aggregates from neurons (blue) bind Toll-like receptor 2 (red) on other cells, leading to internalization of the aggregates and causing death in other neurons, demyelination in oligodendrocytes, and neuroinflammation in microglia. [Courtesy of Seung-Jae Lee, Neuramedy.]
Toll-like receptor 2 (TLR2) has been blamed for bringing α-synuclein aggregates into neurons, as well as for sparking harmful inflammation in microglia (Apr 2020 conference news; Oct 2020 news; Kim et al., 2021). The antibody tomaralimab, developed by Opsona Therapeutics, Ireland, and acquired by the South Korean company Neuramedy, binds TLR2 and blocks its activity. It lowered α-synuclein deposits and gliosis while improving motor abilities in a mouse model of neuronal synucleinopathy (Apr 2022 conference news). In Lisbon, Seung-Jae Lee of Seoul National University, South Korea, extended the findings to multiple system atrophy (MSA).
In this fast-progressing synucleinopathy, aggregates known as glial cytoplasmic inclusions (GCIs) form in oligodendrocytes. As in Parkinson’s disease, dopaminergic neurons die, and motor abilities deteriorate. Lee, who works for Neuramedy, noted that the origin of GCIs has always been mysterious, because oligodendrocytes make very little α-synuclein. The leading hypothesis is that these cells take up α-synuclein aggregates released by neurons.
Supporting this idea, Lee found that cultured human oligodendrocytes internalized α-synuclein oligomers added to their media, triggering GCI formation (Yoon et al., 2020). Similarly, in mice that expressed human mutant A53T α-synuclein only in neurons, phosphorylated α-synuclein showed up in oligodendrocytes, too, indicating it had passed between cells.
Lee theorized that TLR2 was the gateway for α-synuclein entry into oligodendrocytes. In keeping with this, pretreating cultured oligodendrocytes with tomaralimab before adding α-synuclein prevented GCIs, he reported. Likewise, injecting A53T mice with 10 mg/kg tomaralimab weekly from 6 to 10 months of age cut phosphorylated α-synuclein in white matter by half. Tomaralimab also prevented gliosis. Treated mice maintained their grip strength and balance, and they survived as long as did wild-type mice.
Lee next wanted to know how α-synuclein damages oligodendrocytes. RNA-Seq analysis of cultured human oligodendrocytes, as well as postmortem samples from MSA patients, found that glia containing GCIs were likelier than healthy glia to have expression profiles characteristic of immature cells. In particular, they expressed fewer myelination genes. This suggested that α-synuclein triggers oligodendrocytes to de-differentiate, causing demyelination. Treating cultured oligodendrocytes with tomaralimab restored their mature phenotype, Lee found. In mice, treatment maintained healthy myelin.
Targeting TLR2 could have multiple benefits in synucleinopathy, keeping neurons healthy, myelin robust, and gliosis down, Lee concluded. Clinicaltrials.gov lists a planned Phase 1 trial of tomaralimab in healthy volunteers in the U.K. to test safety and pharmacokinetics, but Lee told Alzforum that study has been shelved. Instead, Neuramedy will run the Phase 1 trial in South Korea. Lee said the company will focus first on MSA trials, rather than PD, as the former is faster-progressing and may soon have a PET tracer available (Mar 2022 conference news; Smith et al., 2023).

Disease Marker? In CSF from Parkinson’s patients (red, left) total α-synuclein is only a little lower than in healthy controls (blue). In contrast, nitrated α-synuclein (right) in patient CSF dwarfs that in control CSF. [Courtesy of Sheerin Shahidi-Latham, Nitrase Therapeutics.]
Could Nitrated α-Synuclein Hold the Key to Toxicity?
In contrast to this indirect TLR2 immunotherapy strategy, Sheerin Shahidi-Latham of the biotech Nitrase Therapeutics, Brisbane, California, proposed directly binding α-synuclein—but only its nitrated form. Nitrated α-synuclein was first implicated in neuron damage in PD more than two decades ago, but few have pursued this target (Giasson et al., 2000; Yu et al., 2010; Apr 2011 conference news).
In Lisbon, Shahidi-Latham made a case for nitration being tightly linked to disease. She reported that nitrated α-synuclein (nSyn) was present in cerebrospinal fluid from 50 PD patients, but virtually absent from the CSF of 50 healthy controls (see image above). Moreover, having more than 15 pg/mL nSyn in CSF correlated with having a positive α-synuclein seed amplification assay (Apr 2023 conference news; Aug 2023 conference news). Notably, three PD patients who were below this nSyn threshold were SAA-negative as well, strengthening the link between nSyn and aggregates (see image below). “This points to nSyn having a pathogenic role in PD,” Shahidi-Latham suggested.

Nitrated α-Synuclein and Aggregation. People with more than 15 pg/mL nitrated α-synuclein in their CSF (dotted line) also had positive α-synuclein seed amplification assay (SAA) tests (black), while those with less did not, even if they had been diagnosed with PD. [Courtesy of Sheerin Shahidi-Latham, Nitrase Therapeutics.]
The company generated several antibodies against mouse nSyn, picking the most selective and potent candidate to test. Researchers injected preformed α-synuclein fibrils into the striatum of A53T M83 mice to stimulate α-synulcein aggregation, and waited one week. Then they injected 30 or 100 mg/kg antibody weekly for 12 weeks. The lead antibody candidate dose-dependently suppressed α-synuclein phosphorylated at Ser129, a marker for aggregated protein, and reached statistical significance at the higher dose. By contrast, Roche and Prothena’s α-synuclein antibody prasinezumab had no effect in these mice. Prasinezumab had been negative in a Phase 2 Parkinson’s trial, but had modest benefits on secondary outcomes and in subgroups (Apr 2020 conference news; Apr 2021 conference news).
Shahidi-Latham noted that in Parkinson’s patients, only 3 percent of α-synuclein in the CSF is nitrated. Why, then, would targeting this form work better than binding all forms of the protein? Shahidi-Latham drew a parallel with donanemab, which targets a pyroglutamate, aggregating form of Aβ and showed high efficacy in removing plaque in a Phase 3 trial (May 2023 news; Jul 2023 conference news). By selectively targeting a toxic form, she believes her antibody prevents secreted nSyn from seeding aggregation and promoting the spread of pathology.
The company has now humanized the antibody, and optimized it to make it more potent. That version, NDC-0524, has a half-life in human plasma of 18 days, compatible with monthly dosing. Modeling suggests that a dose of less than 30 mg/kg will reach effective concentrations in people, Shahidi-Latham said. The company plans to take NDC-0524 into Phase 1 next year, using change in CSF nSyn and SAAs as readouts for efficacy.

Breaking Up Toxic Couple. Small-molecule syntacasyn (red dots) binds synaptic protein synapsin III (gray) and detaches it from α-synuclein fibrils (purple), causing fibrils to fall apart. Syntacasyn then encourages binding of synapsin III to physiological α-synuclein, restoring dopamine homeostasis. [Courtesy of Arianna Bellucci.]
Busting Aggregates by Hitting a Synaptic Protein
Small molecules have an advantage for tackling synucleinopathies, since they can enter cells to interact directly with deposits. Arianna Bellucci of the University of Brescia, Italy, debuted one such molecule that takes a unique approach to busting up fibrils—by first making them split up with a co-aggregating synaptic partner. Bellucci noted that α-synuclein loiters in pre-synapses, putting it in the right place to bind synaptic proteins, and that its presence there correlates with neurodegeneration (Schulz-Schaeffer, 2010). She previously reported that α-synuclein interacts with synapsin III in nerve terminals to regulate dopamine release (Zaltieri et al., 2015). Moreover, she detected synapsin III in Lewy bodies, suggesting it plays a role in pathology (Longhena et al., 2018).
To investigate synapsin III’s role, Bellucci overexpressed α-synuclein in the nigrostriatum of wild-type and synapsin III knockout mice. While the wild-types developed α-synuclein aggregates and lost neurons, the synapsin III knockouts were protected (Faustini et al., 2018). Lowering synapsin III also helped mice that already had synuclein pathology. In transgenic mice expressing human truncated α-synuclein, silencing synapsin III in the nigrostriatum broke up existing aggregates. Dopamine release recovered, and neuron number stabilized (Faustini et al., 2022).
This led Bellucci and colleagues to search for small molecules that could interfere with synapsin III binding to α-synuclein fibrils. The monoamine reuptake inhibitor methylphenidate, which binds synapsin III, emerged as the best candidate. MPH is approved to treat attention deficit hyperactivity disorder, a disease associated with synapsin III polymorphisms, Bellucci noted. In synucleinopathy mice, MPH lowered fibril load and improved movement. This effect did not depend on its monoamine transporter activity (Faustini et al., 2020).
The researchers fiddled with MPH’s structure to develop derivatives that bound synapsin III more strongly, while not affecting monoamine transport. They named their lead candidate syntacasyn. Syntacasyn detaches synapsin III from α-synuclein fibrils, causing them to unravel. In computer simulations of protein interactions, it nudges α-synuclein back toward a physiological, α-helical shape, allowing it to interact with soluble synapsin III, Bellucci said. This restores normal dopamine release. In addition, syntacasyn enters the brain well and has a favorable pharmacokinetic profile, Bellucci said. In mice, the compound was not toxic, even at 10 times the effective dose.
The researchers tested syntacasyn in 50-day-old midbrain organoids made from patients with an α-synuclein gene triplication. After 30 days, 200 nM syntacasyn had lowered α-synuclein aggregates by about 80 percent, while maintaining dopaminergic neurons at the same level as in control organoids. Similarly, mice expressing human C-terminally truncated α-synuclein that were treated for four weeks had about one-quarter as much aggregated α-synuclein as untreated mice, and did not lose dopaminergic neurons.
“We can destabilize α-synuclein aggregates by controlling the interplay between α-synuclein and synapsin III,” Bellucci concluded. She told Alzforum that she is planning to test syntacasyn in clinical trials.
Aggregation Inhibitor Heads to Phase 2
Further along the development pipeline is a molecule familiar to Alzforum readers. In Lisbon, Johannes Levin of Ludwig Maximilian University, Munich, presented the latest on the oligomer-busting drug anle138b, now rechristened emrusolmin. In earlier studies in Parkinson’s disease models, this small molecule entered the brain well, bound α-synuclein, and broke up aggregates by changing the protein’s shape (Apr 2011 conference news; Mar 2015 conference news).
Armin Giese at Ludwig Maximilian, who first identified emrusolmin, co-founded the pharma company MODAG GmbH to take the compound into trials (Aug 2014 conference news). The company has completed Phase 1 studies in healthy volunteers and PD patients. Emrusolmin had a half-life of 12 hours, compatible with once-daily dosing, and achieved plasma levels above those shown to be effective in mouse models. Doses up to 300 mg were well-tolerated (Levin et al., 2022).
Levin said the company will next test the drug in MSA. PLP-haSyn mice model MSA by driving α-synuclein expression in oligodendrocytes (Kahle et al., 2002). When fed emrusolmin from two to six months of age, these mice maintained their dopaminergic neurons, mounted little microgliosis, and could balance on a narrowing beam as well as wild-types (Heras-Garvin et al., 2019).
A Phase 2 trial dubbed Topas-MSA will enroll 160 people at early disease stages, when they are still able to walk at least 30 feet unassisted. Participants will take 300 mg emrusolmin or placebo once daily for 48 weeks. The primary outcome will be the modified unified MSA rating scale part 1. The trial will take place in the U.S., France, Italy, Germany, Spain, Israel, and Japan, and will start at the end of this year, Levin said.—Madolyn Bowman Rogers
References
News Citations
- Parkinson's Therapies Seek to Stem Progression
- α-Synuclein Spurs Neuroinflammation Via Microglial LRRK2
- UB-312 Synuclein Vaccine Safe in Controls. Next Up: Parkinson's.
- In First for the Field, α-Synuclein PET. Only for Multiple System Atrophy
- Barcelona: Parkinson’s Treatments on the Horizon
- Synuclein Assay Passes the Sniff Test—What of Other Seeds?
- Finally, a Diagnostic Marker for Lewy Body Disease?
- α-Synuclein Antibody Misses Primary, May Have Signal on Secondaries
- For α-Synuclein Immunotherapy, Is Going Later the Key?
- And Then There Were Three: Donanemab Phase 3 Trial Positive
- Donanemab Data Anchors Upbeat AAIC
- Antibody Against α-Synuclein Looks Safe In Phase 1
- Therapies Take Aim at Tau
Research Models Citations
Therapeutics Citations
Paper Citations
- Kim C, Kwon S, Iba M, Spencer B, Rockenstein E, Mante M, Adame A, Shin SJ, Fields JA, Rissman RA, Lee SJ, Masliah E. Effects of innate immune receptor stimulation on extracellular α-synuclein uptake and degradation by brain resident cells. Exp Mol Med. 2021 Feb;53(2):281-290. Epub 2021 Feb 16 PubMed.
- Yoon YS, Ahn WJ, Ricarte D, Ortiz D, Shin CY, Lee SJ, Lee HJ. Alpha-Synuclein Inclusion Formation in Human Oligodendrocytes. Biomol Ther (Seoul). 2020 Jun 15; PubMed.
- Smith R, Capotosti F, Schain M, Ohlsson T, Vokali E, Molette J, Touilloux T, Hliva V, Dimitrakopoulos IK, Puschmann A, Jögi J, Svenningsson P, Andréasson M, Sandiego C, Russell DS, Miranda-Azpiazu P, Halldin C, Stomrud E, Hall S, Bratteby K, Tampio L'Estrade E, Luthi-Carter R, Pfeifer A, Kosco-Vilbois M, Streffer J, Hansson O. The α-synuclein PET tracer [18F] ACI-12589 distinguishes multiple system atrophy from other neurodegenerative diseases. Nat Commun. 2023 Oct 27;14(1):6750. PubMed.
- Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000 Nov 3;290(5493):985-9. PubMed.
- Yu Z, Xu X, Xiang Z, Zhou J, Zhang Z, Hu C, He C. Nitrated alpha-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS One. 2010;5(4):e9956. PubMed.
- Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol. 2010 Aug;120(2):131-43. PubMed.
- Zaltieri M, Grigoletto J, Longhena F, Navarria L, Favero G, Castrezzati S, Colivicchi MA, Della Corte L, Rezzani R, Pizzi M, Benfenati F, Spillantini MG, Missale C, Spano P, Bellucci A. α-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J Cell Sci. 2015 Jul 1;128(13):2231-43. Epub 2015 May 12 PubMed.
- Longhena F, Faustini G, Varanita T, Zaltieri M, Porrini V, Tessari I, Poliani PL, Missale C, Borroni B, Padovani A, Bubacco L, Pizzi M, Spano P, Bellucci A. Synapsin III is a key component of α-synuclein fibrils in Lewy bodies of PD brains. Brain Pathol. 2018 Jan 13; PubMed.
- Faustini G, Longhena F, Varanita T, Bubacco L, Pizzi M, Missale C, Benfenati F, Björklund A, Spano P, Bellucci A. Synapsin III deficiency hampers α-synuclein aggregation, striatal synaptic damage and nigral cell loss in an AAV-based mouse model of Parkinson's disease. Acta Neuropathol. 2018 Oct;136(4):621-639. Epub 2018 Jul 25 PubMed.
- Faustini G, Longhena F, Masato A, Bassareo V, Frau R, Klingstedt T, Shirani H, Brembati V, Parrella E, Vezzoli M, Nilsson KP, Pizzi M, Spillantini MG, Bubacco L, Bellucci A. Synapsin III gene silencing redeems alpha-synuclein transgenic mice from Parkinson's disease-like phenotype. Mol Ther. 2022 Apr 6;30(4):1465-1483. Epub 2022 Jan 14 PubMed.
- Faustini G, Longhena F, Bruno A, Bono F, Grigoletto J, La Via L, Barbon A, Casiraghi A, Straniero V, Valoti E, Costantino G, Benfenati F, Missale C, Pizzi M, Spillantini MG, Bellucci A. Alpha-synuclein/synapsin III pathological interplay boosts the motor response to methylphenidate. Neurobiol Dis. 2020 May;138:104789. Epub 2020 Feb 4 PubMed.
- Levin J, Sing N, Melbourne S, Morgan A, Mariner C, Spillantini MG, Wegrzynowicz M, Dalley JW, Langer S, Ryazanov S, Leonov A, Griesinger C, Schmidt F, Weckbecker D, Prager K, Matthias T, Giese A. Safety, tolerability and pharmacokinetics of the oligomer modulator anle138b with exposure levels sufficient for therapeutic efficacy in a murine Parkinson model: A randomised, double-blind, placebo-controlled phase 1a trial. EBioMedicine. 2022 Jun;80:104021. Epub 2022 Apr 29 PubMed.
- Kahle PJ, Neumann M, Ozmen L, Muller V, Jacobsen H, Spooren W, Fuss B, Mallon B, Macklin WB, Fujiwara H, Hasegawa M, Iwatsubo T, Kretzschmar HA, Haass C. Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep. 2002 Jun;3(6):583-8. PubMed.
- Heras-Garvin A, Weckbecker D, Ryazanov S, Leonov A, Griesinger C, Giese A, Wenning GK, Stefanova N. Anle138b modulates α-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov Disord. 2019 Feb;34(2):255-263. Epub 2018 Nov 19 PubMed.
External Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.